| Journal of Food Bioactives, ISSN 2637-8752 print, 2637-8779 online |
| Journal website www.isnff-jfb.com |
Original Research
Volume 21, March 2023, pages 62-71
Biomolecular interactions and inhibition kinetics of human soluble epoxide hydrolase by tetrapeptide YMSV
Joy I. Obeme-Nmoma, Raliat O. Abioyea, Toluwase H. Fatokib, Chibuike C. Udenigwea, c, *
aDepartment of Chemistry and Biomolecular Sciences, Faculty of Science, University of Ottawa, Ottawa, ON K1N 6N5, Canada
bDepartment of Biochemistry, Federal University Oye-Ekiti, PMB 373, Oye-Ekiti, Ekiti State, Nigeria
cSchool of Nutrition Sciences, Faculty of Health Sciences, University of Ottawa, Ottawa. ON K1H 8M5, Canada
*Corresponding author: Chibuike C. Udenigwe, School of Nutrition Sciences, Faculty of Health Sciences, University of Ottawa, Ottawa, ON K1H 8M5, Canada. E-mail: cudenigw@uottawa.ca
DOI: 10.31665/JFB.2023.18341
Received: March 17, 2023
Revised received & accepted: March 31, 2023
| Abstract | ▴Top |
Soluble epoxide hydrolase (sEH) contributes to the pathophysiology of neurodegenerative diseases by decreasing the epoxyeicosatrienoic acids/dihydroeicosatrienoic acids ratio and influencing the anti-inflammatory system. Thus, sEH inhibition reduces systemic inflammation, particularly in the brain. This study investigated sEH inhibition by a tetrapeptide, YMSV, and its mechanism of action. Enzyme inhibition kinetics demonstrated that YMSV is a mixed-competitive inhibitor of sEH, with a half-maximal inhibitory concentration (IC50) of 179.5 ± 0.92 µM. Secondary structural analysis of sEH by circular dichroism showed that YMSV decreased the α-helices by 7.7% and increased the β-sheets and random coils by 11.4% and 22%, respectively. Molecular docking simulation indicated that YMSV formed a hydrogen bond with the Asp333 residue of the hydrolase pocket of sEH in addition to the binding of non-active site residues. The findings provide new insights into the mechanism of sEH inhibition by YMSV and its potential as a peptide-based anti-depressant nutraceutical.
Keywords: Soluble epoxide hydrolase (sEH); Enzyme inhibition kinetics; Bioactive peptides; EET; DHET; Biomolecular interaction
| 1. Introduction | ▴Top |
Soluble epoxide hydrolase (sEH) has been studied extensively for its preclinical and clinical importance and implications in several diseases. sEH is involved in the metabolic transformation of epoxides into less active diols. It is predominantly expressed in the liver, brain, kidney, heart, lung, spleen, adrenal, and gut, where it plays a role in systemic inflammation and blood pressure regulation. Within the cell, sEH is found in the peroxisome, membrane, and cytosol (Hollinshead and Meijer, 1988). The 120-kDa homodimer enzyme belongs to the α/β-hydrolase fold superfamily and catalyzes the hydrolysis of epoxides using its C-terminal domain and the hydrolysis of phospholipids using the N-terminal domain (Morisseau and Hammock, 2013). sEH substrates are biologically important epoxides known as epoxyeicosatrienoic acids (EETs), which are derived from arachidonic acid metabolism in the cytochrome P450 pathway. Hydrolysis of EETs by sEH into the less active dihydroeicosatrienoic acids (DHETs) fundamentally affects the overall regulation of epoxy fatty acids (EpFAs) and inflammation in the body (Swardfager et al., 2018). EETs are signaling molecules that are prevalently abundant in the liver, smooth muscles, and blood vessels of other major organs. Their functions in cardioprotection, fibrinolysis, smooth muscle migration, angiogenesis, vasodilation and prevention of inflammation, apoptosis, and oxidation can be attributed to various mechanisms such as decreased TNF-α secretion in monocytic cells, inhibition of ICAM-1, VCAM-1, and E-selectin expression, and obstruction of NF-κB nuclear translocation, which in turn downregulates several enzymes, including inducible nitric oxide synthase (iNOS) (Khan et al., 2013). Consequently, sEH has attracted notable attention due to the wide variety of therapeutic potential shown by EETs, especially in cardiovascular, neurodegenerative, and metabolic diseases (Sun et al., 2021).
Bioactive peptides are a sequence of 2–20 amino acids that have beneficial health-promoting properties, including anti-inflammatory, antioxidant, anticancer, antihypertensive, antimicrobial, and immunomodulatory activities. The biological functions of these peptides make them important candidates for pharmaceutical and therapeutic treatments for various diseases, such as diabetes, hypertension, and neurodegenerative diseases (Udenigwe et al., 2021). Several in vivo and in vitro studies have demonstrated the anti-inflammatory effects of food-derived bioactive peptides. Peptides derived from eggs, milk, and soybean have expressed anti-inflammation by reducing the expressions of TNF-κ, IL-10, IL-6, IL-1β, ICAM-1 and VCAM-1 as well as inhibition of nitric oxide production (Guha and Majumder, 2019). Specifically, bioactive peptides from milk and soy proteins have been reported to inhibit the NF-κB pathway by decreasing the levels of NF-κB in the cell nucleus and increasing the concentration of IκB concentration in the cytosol, which led to Nrf2 activation, and the expression of IL-1ra (interleukin-1 receptor antagonist protein) which is an anti-inflammatory cytokine (Tonolo et al., 2022). Similar mechanisms have been reported for EETs; thus, the bioactive peptides can be applied in improving the inflammation response of the body.
The pharmacophore of many first chemically synthesized SEH inhibitors, such as 12-(3-adamantan-1-yl-ureido) dodecanoic acid (AUDA), involve the incorporation of urea analogues and amide groups to produce a similar configuration as the epoxide while employing an arachidonic acid backbone as the model structure. Multitarget inhibitors of sEH and other enzymes and receptors involved in the inflammatory pathway, such as COX-2, FAAH, and PPAR, have been discovered (Iyer et al., 2022; Sun et al., 2021). The dual sEH inhibitor t-TUCB has been described as a poor inhibitor of FAAH (Kodani et al., 2018); PTUPB also interacts with the histidine and tyrosine residues of the COX-2 active site by hydrogen bonding (Hwang et al., 2011; Zhang et al., 2014); AUBPC and TPUBPC are effectors designed to inhibit sEH while activating PPARα/γ (Buscató et al., 2012). Naturally derived sEH inhibitors have also been identified in various plants, including flavonoids from Capsicum Chinese jacq. Cv. Habanero (Kim et al., 2019), phenolic compounds from Vietnam’s Passiflora edulis seeds (Cuong et al., 2019), anthraquinone and stilbene derivatives from Rheum undulatum (Jo et al., 2016), as well as triterpenes and phenylpropanoid derivatives from Lycopus lucidus roots (Han et al., 2021). However, these natural sEH inhibitors have poor pharmacokinetics properties, including low water solubility, absorption in the gastrointestinal tract, and bioaccessibility (Lee et al., 2020). In addition to the amide group pharmacophore, many bioactive peptides are relatively stable with good pharmacokinetics properties, and some have the ability to penetrate the blood-brain barrier (BBB), thus the possibility of their development as sEH inhibitors and anti-depressive nutraceuticals. This study was conducted with the goal of determining the mechanisms of inhibition and biomolecular interactions of tetrapeptide YMSV as an inhibitor of soluble epoxide hydrolase.
| 2. Materials and method | ▴Top |
2.1. Materials and reagents
Human soluble epoxide hydrolase and 3-phenyl-cyano(6-methoxy-2-naphthalenyl) methylester-2-oxirane acetic acid (PHOME) were purchased from Cayman Chemical (Ann Arbor, Michigan, USA). Peptide YMSV was synthesized with a purity of over 95% by GenScript Inc. (New Jersey, USA). Bovine serum albumin (BSA), bis-Tris-HCl, 6-methoxy-2-naphthaldehyde (6M2N), and dimethyl sulfoxide (DMSO) were purchased from MilliporeSigma (Oakville, ON, Canada). All the chemical reagents are analytical grade and used without further purification.
2.2. sEH inhibition assay
The in vitro inhibition of soluble epoxide hydrolase by YMSV peptide was conducted by a fluorometric assay using 3-phenyl-cyano(6-methoxy-2-naphthalene) methyl ester-2-oxirane acetic acid (PHOME) as previously described with some modifications (Abis et al., 2019a; Kim et al., 2019). Briefly, 50 µL of 16 ng/mL human sEH in the reaction buffer (25 mM bis-Tris-HCl buffer, pH 7.0, containing 0.1% BSA), and 50 μL of peptide YMSV (6.25–250 μM, in reaction buffer) were mixed in a black 96-well plate, followed by the addition of 50 μL of the buffer containing 5 μM PHOME. The appearance of the fluorometric product, 6-methoxy-2-naphthaldehyde (6M2N) was then measured as relative fluorescence unit (RFU) for 1 h using the Spark multimode microplate reader (Tecan, Stockholm, Sweden), with the following setup: excitation wavelength 330 nm, emission wavelength 460 nm, detection every 45 s, gain 750, and 30 °C constant temperature. The reaction rate (RFU/min) was calculated as:
The IC50, the concentration of the inhibitor that reduced the activity of sEH by 50%, was determined using the linear regression obtained from the curve of the percent inhibition vs. inhibitor concentration.
2.3. Enzyme inhibition kinetics studies
The kinetics of sEH-catalyzed conversion of PHOME to 6-methoxy-2-naphthaldehyde was studied within the first 30 mins of the enzyme assay in the absence and presence of 25–250 µM YMSV at 1.25, 2.5, 5, 10 and 20 µM PHOME, a range previously reported for sEH kinetics studies (Jo et al., 2016). The mode of inhibition was determined by Lineweaver-Burk plot and enzyme kinetics parameters (Km and Vmax) were determined from Lineweaver-Burk plot using GraphPad Prism version 9.1.2 for Windows (GraphPad Software, La Jolla, CA, USA). To test for the effect of substrate (PHOME) on sEH inactivation in the presence and absence of YMSV, the kinetics assay was carried out for 6 h to determine kinetic inactivation parameters (P∞ and apparent inactivation rate constant, A). The concentration of the product (6M2N) released by sEH was determined using a standard curve of 6M2N.
2.4. Circular dichroism spectroscopy
The effect of YMSV on the secondary structure of human sEH was examined using the Jasco J-715 circular dichroism spectrophotometer (Jasco Corp., Tokyo, Japan). Measurements were performed at a human sEH/YMSV molar ratio of 1:1 with a final human sEH concentration of 64 ng/mL and YMSV concentration of 250 µM in 100 mM phosphate buffer (pH 7.4). The final reaction volume of 250 µL was incubated for 5 min at 30 °C. Measurements were carried out at room temperature in nitrogen gas using a quartz cuvette with a path length of 1 mm. Three scans were recorded and averaged with the wavelength ranging from 185 to 280 nm at a scanning speed of 50 nm/s. Phosphate buffer was used as blank to perform baseline subtraction of the spectral output, and the result was translated into mean residue ellipticity (deg cm2 dmol−1) using a mean residue weight of 64 kDa and a human sEH concentration of 0.025 mg/mL. All data processing was carried out using CDToolX (Miles and Wallace, 2018). The data acquired were plotted using GraphPad Prism version 9.1.2 for Windows (GraphPad Software, La Jolla, CA, USA). Bestsel, a software for CD fitting, was used to calculate the secondary structure contents using a wavelength range of 190–250 nm (Micsonai et al., 2018).
2.5. Molecular docking simulation
Molecular docking studies were carried out with human sEH and YMSV. Briefly, human sEH structure (PDB ID: 3otq) was obtained from www.rcsb.org/pdb. YMSV sequence was converted to SMILES (Simplified Molecular Input Line Entry Specification) format using PepSMI (https://www.novoprolabs.com/tools/convert-peptide-to-smiles-string) and the SMILES string was converted to PDB format using SwissTargetPrediction (http://www.swisstargetprediction.ch/) (Daina et al., 2017). PyMol software was used to prepare the protein by the removal of water and existing ligands. Both YMSV (ligand) and human sEH protein were prepared for docking using AutoDock Tools (ADT) v1.5.6 (Morris et al., 2009) at default settings, and the output file was saved in pdbqt format. Active site amino acid residues of the enzyme were obtained from the literature (Schiøtt and Bruice, 2002) and used to define the docking parameters: center grid box (78.369 × −7.222 × 65.775 points), size (70 × 70 × 70 points), and spacing (0.375 Å). The molecular docking program AutoDock Vina v1.2.3 (Eberhardt et al., 2021; Trott and Olson, 2010) was employed for the docking experiment (Rentzsch and Renard, 2015). After docking, close interactions of binding of the target with the ligands were analyzed and visualized using PyMol and ezLigPlot (Tao et al., 2019).
2.6. ADME analysis
The ADME/Tox properties of peptide YMSV and sEH inhibitor, AUDA, were analyzed and compared. SMILES strings of the peptide were obtained using PepSMI (https://www.novoprolabs.com/tools/convert-peptide-to-smiles-string) while that of AUDA was obtained from the NCBI PubChem Compound database (http://www.pubchem.ncbi.nlm.nih.gov/compound). Pharmacokinetics and drug-likeness were determined using SwissADME, https://www.swissadme.ch/index (Daina et al., 2017), which predicts the absorption, distribution, metabolism, and excretion properties of compounds.
2.7. Statistical analysis
Enzyme inhibition experiments were done in triplicate and data were presented as mean ± standard deviation. Statistical analysis was performed with GraphPad Prism version 9.1.2 for Windows (GraphPad Software, La Jolla, CA, USA) using Tukey’s or Dunnett’s multiple comparisons tests with a significance level set at p < 0.05.
| 3. Results and discussion | ▴Top |
3.1. sEH reaction kinetics and inhibition mode of YMSV
Previous studies have shown that compounds containing urea or amide backbones are excellent and stable inhibitors of human and mouse sEH (Sun et al., 2021). This feature is present in peptides, making them strong sEH inhibitor candidates. Screening of several peptides from our library for sEH inhibition resulted in the selection of YMSV for further studies. YMSV occurs naturally in food proteins, e.g., castor bean (Ricinus communis) protein disulfide-isomerase (f181-184; accession no. Q43116 (PDI_RICCO). Enzyme assays with YMSV (Figure 1a) showed a decrease in the sEH reaction rate (Figure 1b). Concentration-dependent inhibition of sEH by YMSV was apparent at inhibitor concentrations of 25 to 250 µM, with negligible effect at lower concentrations (Figure 1c). The half maximal inhibitory concentration (IC50) was estimated to be 178.5 ± 0.92 µM. A recent study with corn gluten-derived tripeptides WEY, WWY, WYW, YFW and YFY showed lower sEH inhibiting IC50 values ranging from 55.41 to 96 µM (Dang et al., 2022). The higher YMSV IC50 value could be attributed to its larger size and length, compared to the tripeptides, as the size of the latter could facilitate binding to sEH, or specific favorable binding conformation. Molecular size plays an important role in sEH inhibition since the narrow cavity of the enzyme active site might limit access to large and bulky inhibitors (Dang et al., 2022). The aromatic amino acid residues can readily participate in hydrogen bonding because of their -OH and -NH functional groups, and as nucleophiles at the enzyme active site (Scheiner et al., 2002). The tripeptides containing aromatic residues showed high sEH inhibitory activity up to 70% at 100 µM concentration, especially WEY and WWY, which had the lowest IC50 values (Dang et al., 2022). Notably, the tripeptides have a common aromatic amino acid residue, tyrosine, as YMSV, suggesting a similarity in their interactions with sEH.
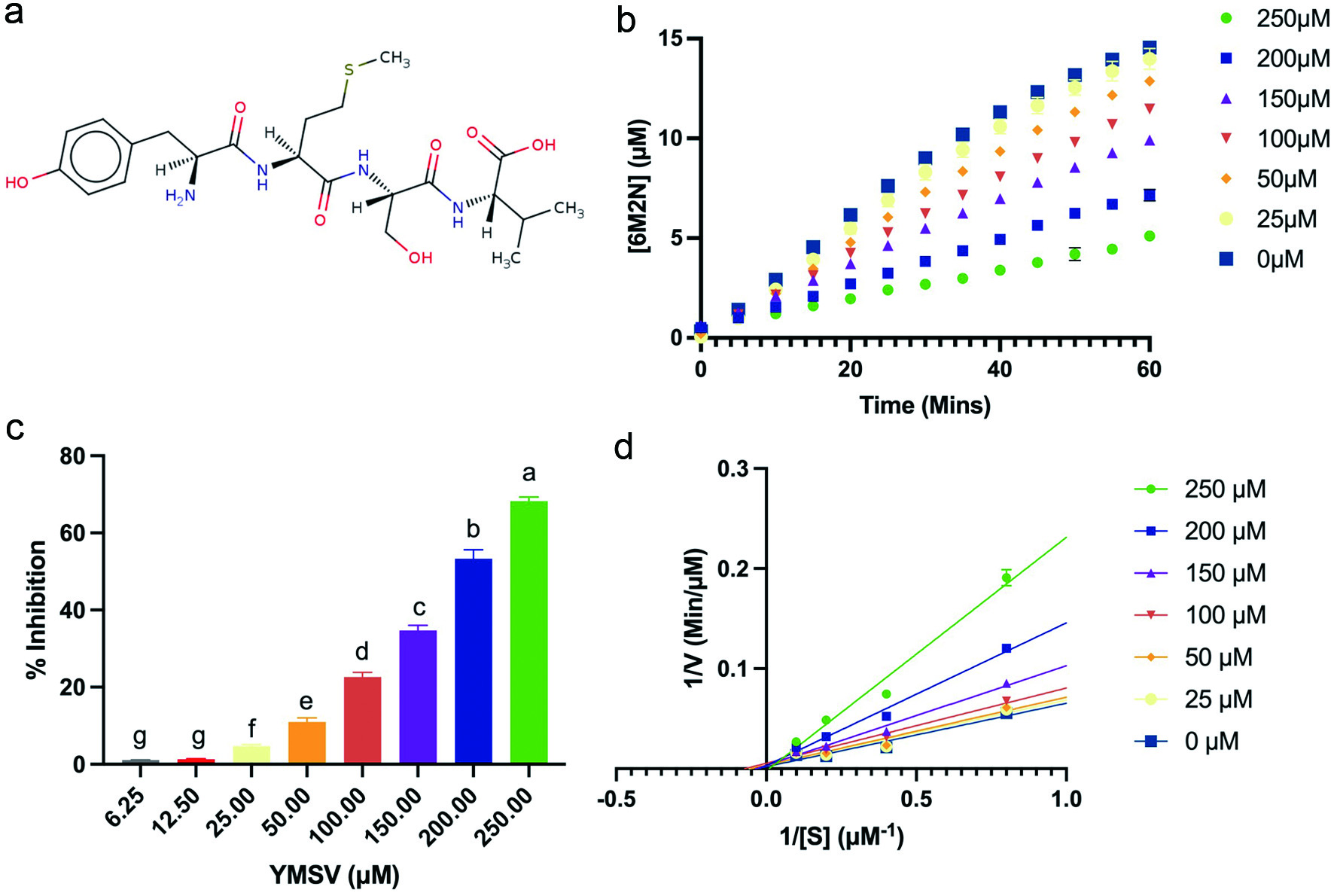 Click for large image | Figure 1. Effect of YMSV on the activity of soluble epoxide hydrolase (sEH). (a) Structure of peptide YMSV; (b) plots of the product (6M2N) formed at constant [S] (5 µM PHOME) over time at different concentrations of the inhibitor, YMSV; (c) concentration-dependent sEH inhibitory activity of YMSV; (d) Lineweaver-Burk double reciprocal plot of sEH at different YMSV concentration. |
A standard curve was prepared using 6M2N to determine the amount of product formed by sEH and the peptide inhibitory effects after 60 min. The rate of the product formed over time at 5 µM substrate and varying concentrations of YMSV is shown in Figure 1b. The concentration of YMSV is inversely correlated with the concentration of the product formed, with a 68% decrease at the highest YMSV concentration.
Lineweaver-Burk plots were used to determine the mode of sEH inhibition by YMSV. Double reciprocal plots of sEH-catalyzed reactions in the presence and absence of YMSV are shown in Figure 1d. The Km value of sEH activity without YMSV was determined to be 3.514 ± 0.02 µM. At lower inhibitor concentrations, the Km values were similar to that of the uninhibited enzyme, indicating that sEH had preferred binding affinity for its substrate, although significant inhibition was observed at these concentrations (Figure 1c). This implies that YMSV likely interacted with the enzyme at other regulatory sites or the enzyme-substrate complex, leading to inhibitory activities. However, higher concentrations of the inhibitor increased the Km of the enzyme. This implies that YMSV was able to competitively bind at sEH active site in place of the substrate, signifying a reduction of substrate affinity. Based on the Lineweaver-Burk plots at varying peptide concentrations, YMSV was shown to be a predominantly competitive inhibitor of sEH (Figure 1d). However, the small but notable changes to the Vmax of the enzyme (Table 1) suggest it to be a mixed-competitive inhibitor. This inhibition mode is similar to that reported for martynoside, a phenylpropanoid isolated from the root of Lycopus lucidus (Han et al., 2021).
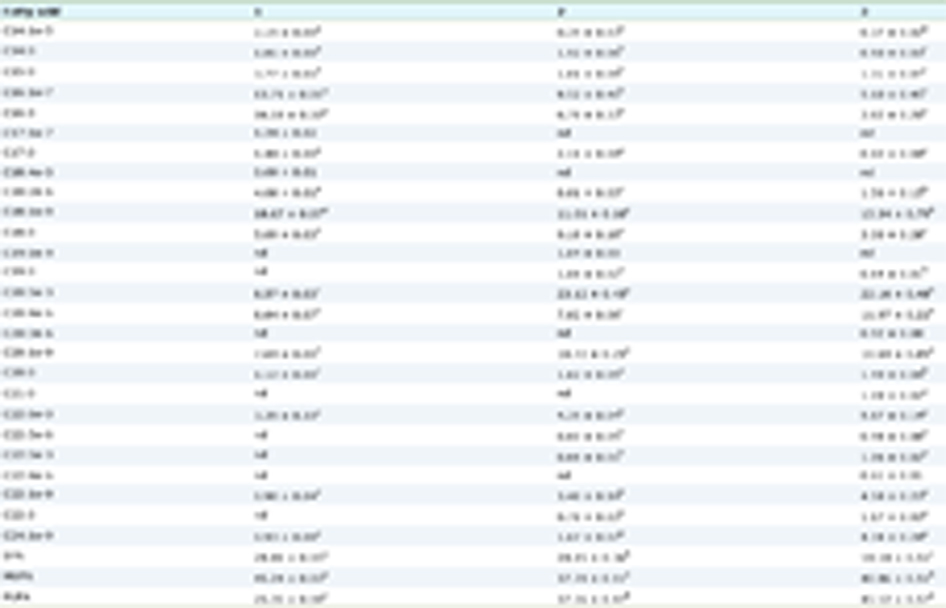 Click to view | Table 1. Kinetics parameters of sEH catalyzed reaction in the absence and presence of different concentrations of the YMSV peptide |
3.2. Dependence on substrate concentration for product formation
Product formation by sEH increased with time in a substrate-dependent manner over the concentration range of YMSV (Figure 2). In addition, the [S] influenced the value of P∞ which is the concentration at which the product remains constant. The concentration of P∞ was inversely proportional to [S]. It is also expected that P∞ would decrease in the presence of the inhibitor given that product formation is reduced in a concentration-dependent manner.
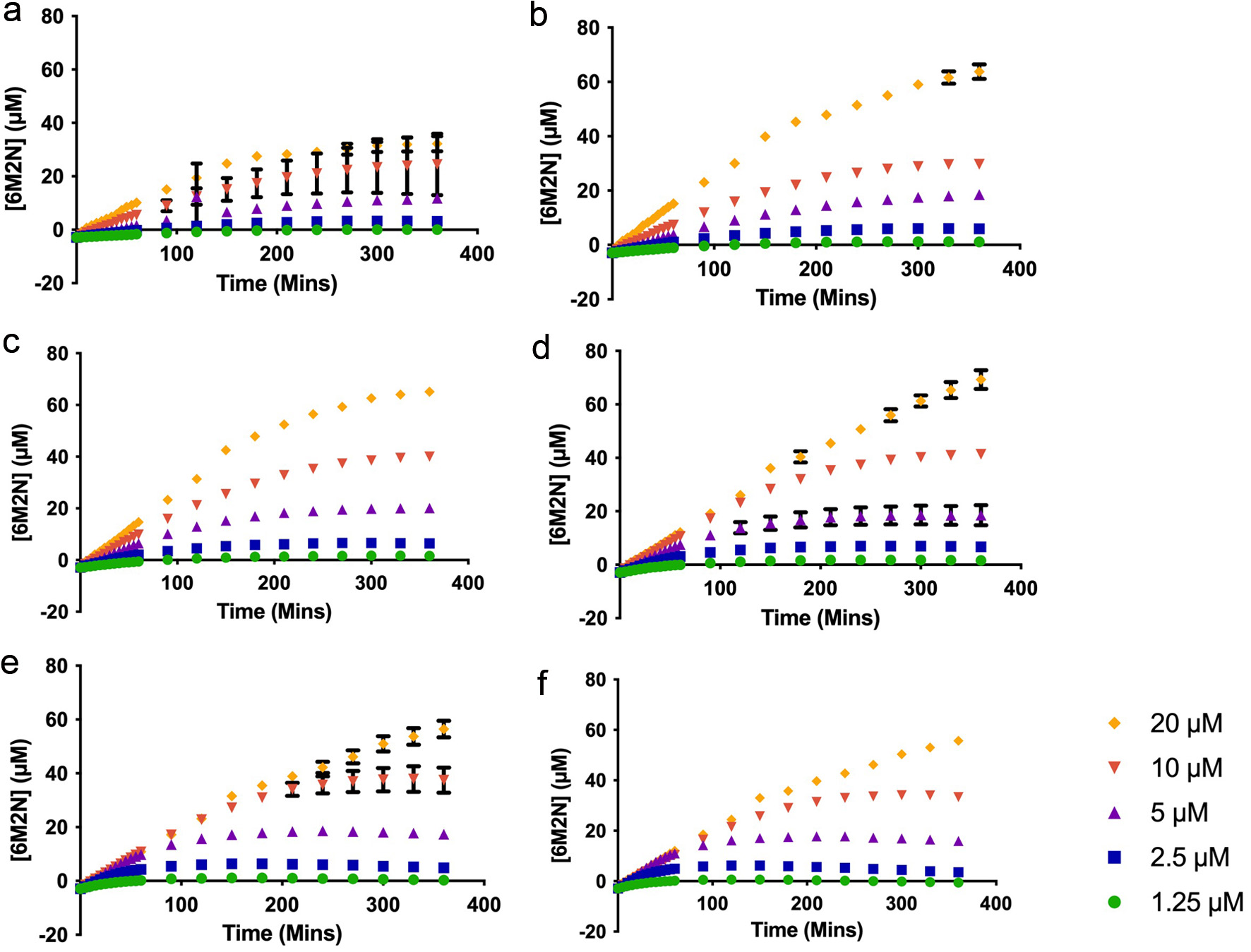 Click for large image | Figure 2. Plots of the product (6M2N) formed by human soluble epoxide hydrolase at the substrate (PHOME) concentrations of 1.25–20 µM in the presence of (a) 250 µM (b) 200 µM (c) 150 µM (d) 100 µM (e) 50 µM (f) 25 µM inhibitor (YMSV). |
As shown in Figure 3, an increase was observed in P∞ as the concentration of YMSV increased from 25–100 µM, after which P∞ started decreasing as more YMSV was added to the enzyme reaction at 10 and 20 µM PHOME. This trend was not observed at lower [S] of 1.25–5 µM PHOME where there was no change in P∞ across all concentrations of YMSV used. This is because most of the substrates were converted into products, resulting in similar P∞ across all the inhibitor concentrations. In addition, lower [S] makes it easier for YMSV to outcompete the substrate for binding to sEH active site, allowing for higher inhibition and reduced P∞. Based on this mechanism, at higher [S] (>10 µM), PHOME would overcome the inhibitory effects of YMSV at lower inhibitor concentrations (<100 µM), resulting in a steady increase in P∞. This suggests that higher [S] promoted binding between the enzyme and substrate, thereby favoring product formation instead of enzyme inhibition by YMSV. The higher concentrations of YMSV resulted in a decrease in P∞, confirming that sEH inhibition occurred by partly decreasing its affinity for the substrate.
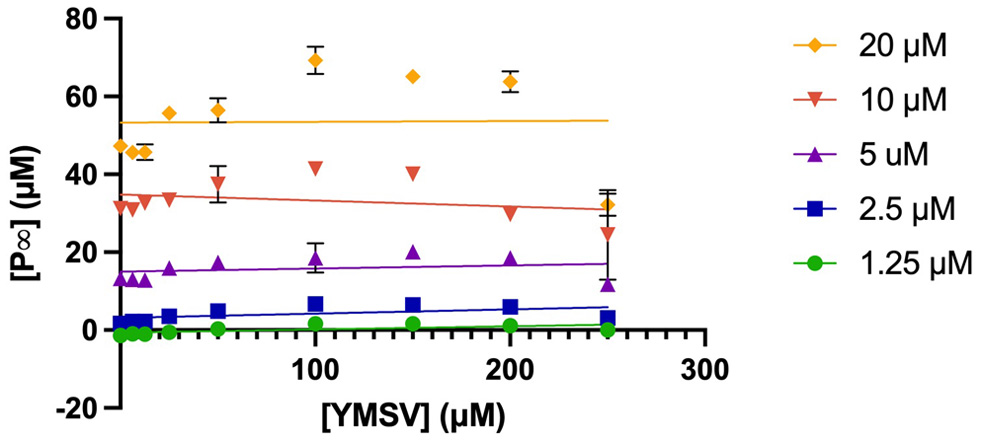 Click for large image | Figure 3. Plots of P∞ against varying concentrations of the inhibitor, YMSV with changing [S] (PHOME = 1.25−20 µM). |
3.3. Apparent inactivation rate constant, A and the protective effect of substrate on inactivation
The apparent inactivation constant, A is obtained by taking the slope of the plot of ln([P]∞ − [P]) vs. time. The plots showed a linear regression with slopes that were dependent on inhibitor concentration (Figure 4). At the lowest concentration of YMSV, the first-order kinetics with negative slopes was observed at lower [S] (<5 µM), while zero-order kinetics was observed at high [S] (>10 µM) (Figure 4a). A higher inhibitor concentration changed the kinetics closer to zero order at all [S] (Figure 4b).
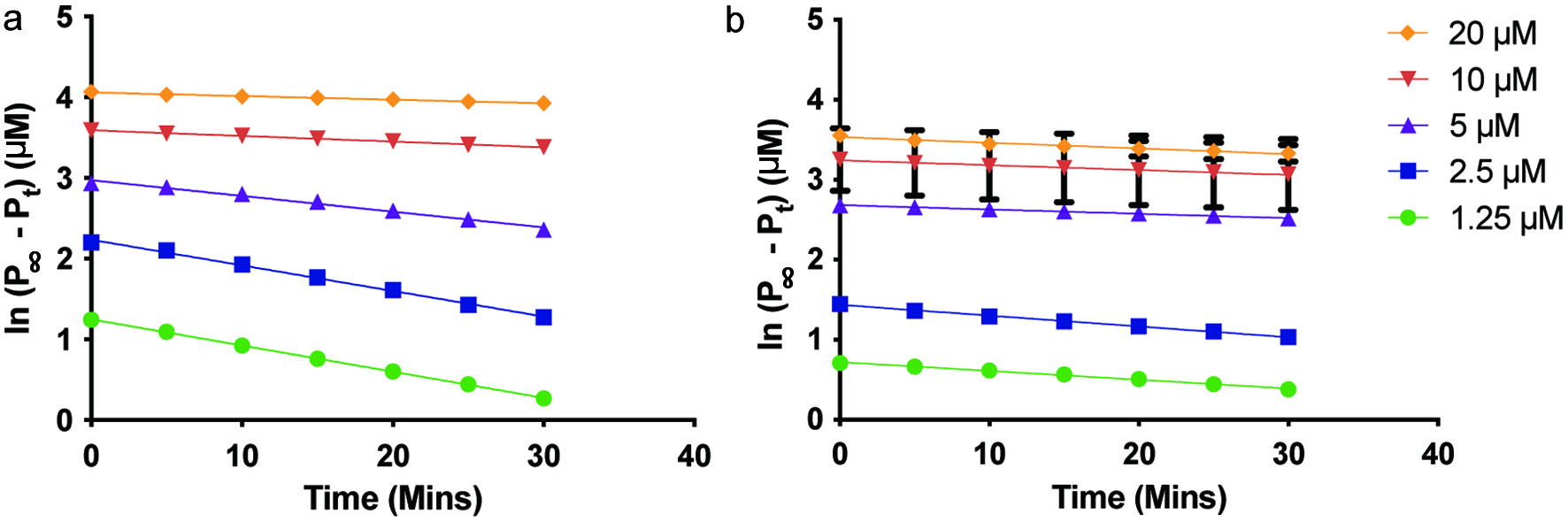 Click for large image | Figure 4. Plots of ln([P]∞ − [P]) vs. time to determine the apparent inactivation rate constant of soluble epoxide hydrolase in the presence of (a) 25 µM YMSV, and (b) 250 µM YMSV. |
A plot of the apparent inactivation rate constant, A against [S] shows the protective effect a substrate may have on an enzyme (Figure 5). The plot derived from the 1/A vs. [S] showed no change in A at [S] below 5 µM, followed by a gradual increase in A as [S] increased in the absence and at low concentration (25 µM) of YMSV. However, at a high concentration of YMSV (250 µM), A increased below [S] of 5 µM, followed by a decrease in A thereafter (Figure 6). No effect on A at low [S] indicates the lack of protective effects by the substrate, thus A is independent of [S] at the lowest YMSV concentration, and to a lesser extent at 250 µM YMSV. This similarity reflects the minimal inhibition that occurred at low inhibitor concentrations. A started to increase with increasing [S], in the presence of 25 µM inhibitor, suggesting the loss of protective effects by the substrate. At 250 µM YMSV, the opposite effect was observed where A increased at [S] >5 µM, suggesting protective effects of the substrate. Taken together, the concentration-dependent effects of the inhibitor on the protective effects of the substrate are apparent.
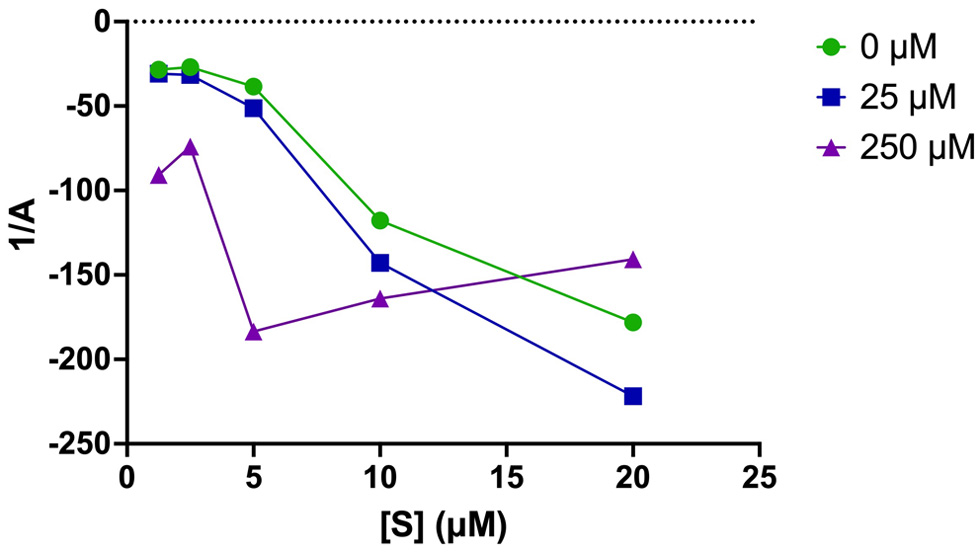 Click for large image | Figure 5. Plots of the reciprocal of apparent inactivation rate constant (A) vs. [S] in the absence and presence of different concentrations of inhibitor (25 and 250 µM YMSV). |
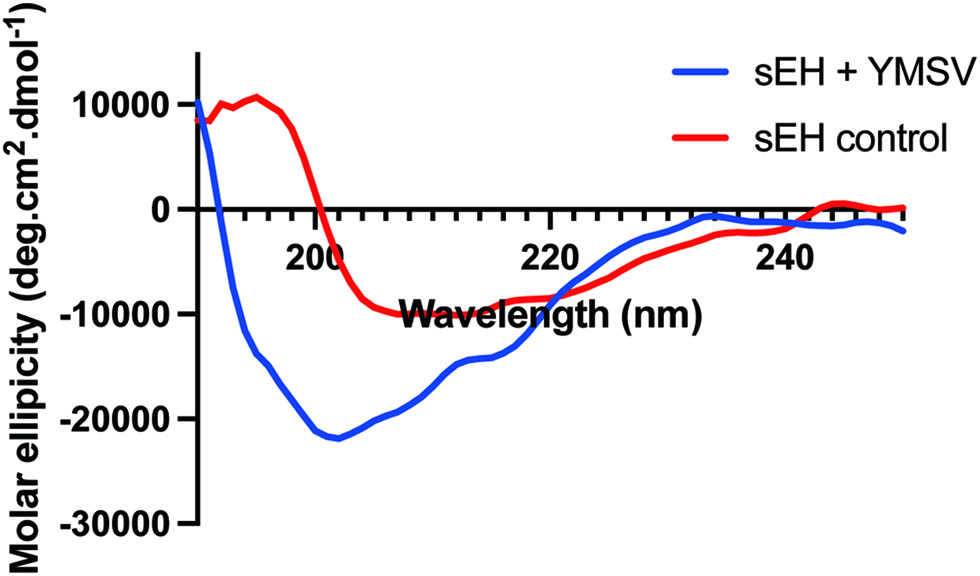 Click for large image | Figure 6. Circular dichroism spectra of soluble epoxide hydrolase in the absence (sEH control) and presence (sEH+YMSV) of the inhibitor peptide. |
3.4. Effect of YMSV on the secondary structure of sEH
Circular dichroism was used to evaluate the effect of YMSV on the secondary structure of sEH toward understanding the potential structural mechanism of inhibition. The pattern of CD spectra obtained for the native sEH is similar to previously reported data (Abis et al., 2019b; de Oliveira et al., 2016). The spectra of sEH control showed the appearance of an absorption maximum at ∼196 nm, a broad negative band with a peak at 207 nm, and a slight shoulder at 222 nm. This absorption pattern suggests the presence of some α-helical content in the secondary structure, the appearance of a broad positive peak from 192–196 nm as well as the negative peak between 210 nm and 220 nm indicating the presence of β-sheet content. As shown in Table 1, the Bestsel program indicated the presence of 23.7% α-helix, 36.1 % anti-parallel β-sheets, 20.1 % parallel β-sheets, and 20.1% turns in sEH control. The presence of YMSV resulted in the disappearance of the strong positive peak with a concomitant broadening of the strong negative peak ranging at 200–235 nm, with a shift in the negative peak and shoulder to 202 and 226 nm, respectively (Figure 6). Analysis with Bestsel indicated that sEH, in the presence of YMSV, contained 16% α-helix, 47.5% anti-parallel β-sheets, 14.4% turns, and 22% others. The addition of YMSV altered the secondary structure contents illustrated by the decrease in α-helical and turns contents and complete loss of parallel β-sheets, some of which may have been converted into the more stable anti-parallel β-sheets. The undefined secondary structures observed in the presence of YMSV could be random coils, suggesting that the inhibitor-induced sEH protein unfolding.
3.5. Molecular docking showing YMSV complexation with sEH
Molecular docking was used to examine the protein-ligand interaction and mechanism by which YMSV inhibited human sEH. The two molecular simulations carried out include active site and blind docking. At sEH active site, the C-terminal hydrolase pocket includes two tyrosine resides, Tyr381 and Tyr465, and a catalytic triad, Asp333-Asp495-His523, which is situated right across from the hydrolase pocket. The carboxylic acid of Asp333 forms an ester bond with the epoxide, which is an important step in epoxide hydrolysis. Due to the nucleophilic nature of Asp333, it can accept hydrogen atom from the amino group (-NH) of the inhibitor peptide to form a hydrogen bond with its oxygen atom. This is the rationale for the competitive inhibition mechanism exhibited by urea- and amide-based sEH inhibitors (Shen and Hammock, 2012). YMSV docking at the active site showed multiple interactions with the sEH, including hydrogen bonding and π-π stacking (Figure 7a). The amino group (NH3 +) at the N-terminal of YMSV was predicted to donate a hydrogen atom to Asp335 in the active site, which corresponds to Asp333 of the catalytic triad. The formation of this hydrogen bond (2.043 Å) confirmed that YMSV could be a competitive inhibitor for sEH. The tyrosine residue of YMSV was also able to have π-π stacking interaction with the Trp 336A. The hydroxymethyl group of Ser and the C-terminus carboxyl group of YMSV participated as ligand acceptors from Asn366A and Ser374A residues, respectively (Figure 7a). Additional interactions between YMSV and side chains (Asn378A, Asn472A, Trp473A, Tyr383A, Tyr446A, Gln384A, Ile363A, Phe381A, Tyr343A, Met339A, Leu499A and Met503A), backbone amino acids (Pro364A), and backbone side chain residues (Pro371A and Met469A) were also observed (Figure 7a). Similarly, competitive inhibition was previously reported for methyl rosmarinate, dimethyl lithospermate and 9″ methyl lithospermate, with similar interacting amino acid residues in sEH as that of AUDA (Han et al., 2021). The binding affinity of YMSV at the active site of sEH was predicted to be −7.669 kcal.mol−1.
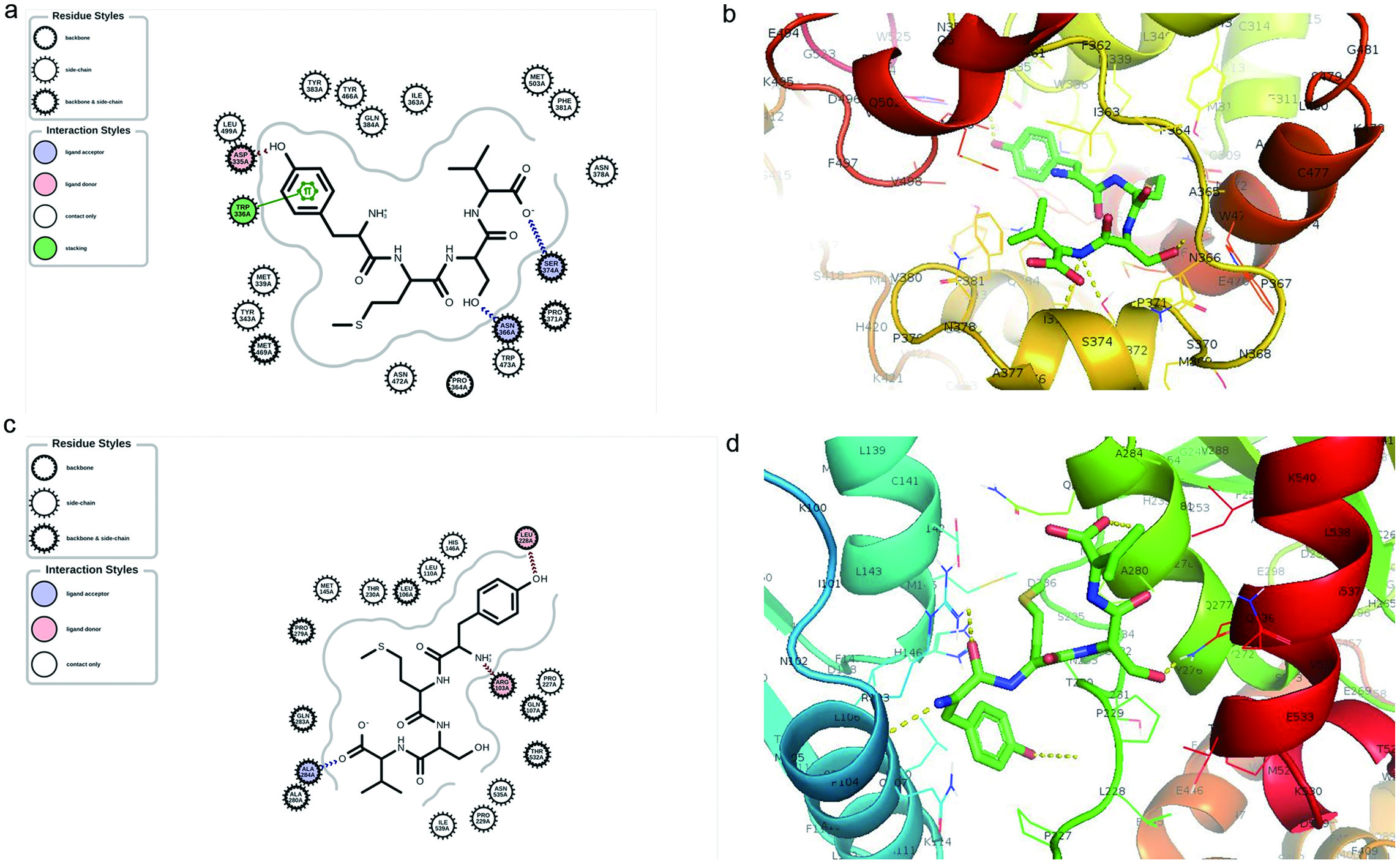 Click for large image | Figure 7. Molecular docking simulation showing (a) 2D and (b) 3D binding interaction of YMSV peptide and the active site of human soluble epoxide hydrolase (sEH), (c) 2D and (d) 3D binding interaction of YMSV peptide and human sEH upon blind docking. |
The blind or free docking of YMSV with sEH showed that the hydroxyl group of Tyr and the amino group at the N-terminal of YMSV acted as a ligand donor to Leu228A and Arg103A residues while the carboxyl group of the C-terminus served as a ligand acceptor for Ala284A residue (Figure 7c). YMSV also interacted with other side chains (Leu110A, Thr230A, His146A, Asn535A, Met145A, Pro229A, Pro227A, and Ile539A) and backbone side chain residues (Leu106A, Pro279A, Gln283A, Ala280A, Thr532A, and Gln107A) of human sEH (Figure 7c). The binding affinity was predicted to be −6.612 kcal.mol−1 (Figure 7d). These findings support the inhibition kinetics results that showed mixed-competitive inhibition.
The previous study with corn gluten tripeptide WEY, a mixed-competitive inhibitor, reported a binding affinity of −10.3024 kcal.mol−1 for the sEH active site (Dang et al., 2022), which was lower that the binding affinities of YMSV from both the active site and blind docking. This implies that YMSV has a lower binding affinity for sEH compared to the tripeptide. Nonetheless, the binding affinity and number of interactions of YMSV predicted at the active site were more favorable than those obtained from blind docking. This shows that YMSV has a higher probability of binding sEH at its active site than other binding sites on the enzyme. In addition, the bond lengths of the hydrogen bonds that were generated at the regulatory and active sites were adequate (Fakhar et al., 2022).
3.6. Comparative in silico drug-likeness of the inhibitors
The computational analysis of the pharmacokinetics and drug-likeness of molecules is a cheaper, faster and non-invasive tool used to predict the behavior of molecules in biological systems (Daina et al., 2019). The SwissADME software was used to predict the ADME (absorption, distribution, metabolism, excretion, and toxicity) potential, which provides the pharmacokinetics, pharmacochemistry, drug-likeness, lipophilicity, and solubility parameters (Daina et al., 2017). The drug-likeness parameters of YMSV were compared to that of AUDA, a widely used strong sEH inhibitor. As shown in Table 2, YMSV is a larger molecule than AUDA; however, it is a small molecule with a molecular weight <500 Da, which is a prerequisite for drug-likeness (Daina et al., 2017, 2019; Ranjith and Ravikumar, 2019). Solubility and lipophilicity are crucial parameters in pharmacokinetics as they describe the interaction of molecules within aqueous and lipid environments. YMSV had a solubility scale Log S value of 0.23, indicating that it is highly soluble compared to AUDA (Log S, −4.98), which can be classified as moderately soluble. AUDA also had a higher CLogP value than YMSV, which has a negative value, indicating a higher affinity for the aqueous phase as confirmed by the ESOL prediction. Compounds having a logP higher than 1 or less than 4 are often thought to have suitable physicochemical and ADME properties required for oral drug intake (Arnott and Planey, 2012). Also, high lipophilicity is characterized by poor solubility, high metabolic rate, and poor absorption, making the compounds potentially harmful as they can accumulate in the body and become toxic (Liu et al., 2011). Most identified sEH inhibitors act by binding to the active site, but their low water solubility impedes their clinical use for the treatment of diseases related to sEH and the inflammatory pathway (Sun et al., 2021). Consequently, a few sEH inhibitors, such as 1,3-disubstituted urea and amide-based inhibitors, have been designed with improved solubility (Kim et al., 2004, 2005). Apart from solubility, these inhibitors are cumbersome in size and, even when soluble in the bloodstream, may not be able to escape biotransformation in the gastrointestinal tract or by the liver enzymes, thereby reducing their bioavailability (Sun et al., 2021). YMSV is particularly susceptible to such modifications, hence its low GIA (Table 2). A previous study demonstrated the bioavailability of YMSV may be reduced due to its high hydrophobicity and lack of charges, which led to its weak mucin-binding properties and low mucus permeation (Sun et al., 2021). YMSV does not inhibit cytochrome P450 3A4 and is a P-glycoprotein substrate; thus, it can be rapidly metabolized and removed from cells in the body which suggests its suitability for human consumption. In addition to sEH inhibition, YMSV was previously reported to inhibit the fibrillation of islet amyloid polypeptide, a factor that contributes to the development of type 2 diabetes (Abioye et al., 2022). Thus, YMSV has promise as a multifunctional nutraceutical compound.
Click to view | Table 2. In silico absorption, distribution, metabolism, and excretion (ADME) profile of the YMSV peptide and AUDA generated using SwissADME |
| 4. Conclusion | ▴Top |
In this study, YMSV was identified as a new mixed-competitive inhibitor of sEH. The interaction of YMSV with sEH resulted in the loss of α-helical and β-sheet content of the enzyme, in favor of unstructured random coil conformation. Molecular docking reveals a myriad of interactions within the catalytic site and backbone residue side chains, which support the proposed mode of inhibition. Predictions of ADME indicate that some of the pharmacokinetics and drug-likeness properties of YMSV are better than AUDA, although the peptide is predicted to have low gastrointestinal absorption and oral bioavailability. Future research should include the development of processing methods to release the bioactive peptide from its parent proteins as well as in vivo physiological bioactivity and pharmacokinetics assessments. Taken together, this study provides important insight into the mechanism by which peptides inhibit sEH activity toward future applications as nutraceuticals for the management of inflammatory diseases.
Acknowledgments
J.I.O. is a recipient of the University of Ottawa Nutrition and Mental Health Scholarship. R.O.A. is a recipient of the Vanier Canada Graduate Scholarship.
This research was supported by the University Research Chair Program of the University of Ottawa, and the Natural Sciences and Engineering Research Council of Canada (NSERC), RGPIN-2018-06839.
Conflict of interest
The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
| References | ▴Top |