| Journal of Food Bioactives, ISSN 2637-8752 print, 2637-8779 online |
| Journal website www.isnff-jfb.com |
Review
Volume 3, September 2018, pages 87-94
Potential effects of natural dietary compounds on trimethylamine N-oxide (TMAO) formation and TMAO-induced atherosclerosis
Pei-Yu Chena, Chi-Tang Hob, Fereidoon Shahidic, Min-Hsiung Pana, *
aInstitute of Food Science and Technology, National Taiwan University, Taipei 10617, Taiwan
bDepartment of Food Science, Rutgers University, New Brunswick, NJ 08901-8520, USA
cDepartment of Biochemistry, Memorial University of Newfoundland, St. John’s, NL, A1B 3X9, Canada
*Corresponding author: Dr. Min-Hsiung Pan, Institute of Food Science and Technology, National Taiwan University, No.1, Section 4, Roosevelt Road, Taipei 10617, Taiwan
DOI: 10.31665/JFB.2018.3151
Received: August 31, 2018; Revised received & accepted: September 10, 2018
| Abstract | ▴Top |
Cardiovascular disease (CVD) is the leading cause of death worldwide. Recently, trimethylamine N-oxide (TMAO) is identified to be highly associated with CVD development and exacerbates atherosclerosis by several mechanisms. TMAO is a gut microbiota-dependent metabolite formed from dietary quaternary amines, mainly choline and carnitine. These trimethylamine (TMA)-containing compounds are first converted to TMA by enzymes in gut microbiota and subsequently metabolized by the host hepatic enzymes to TMAO. As the microbiome is the source of TMAO, administration of broad spectrum antibiotics shows marked decrease in TMAO levels. However, antibiotics may possess many possible undesirable side effects and chronic treatment consideration effects of antibiotic resistance in bacteria. Thus, studies have focused on the alternative strategies, including use of natural dietary compounds to reduce elevated TMAO levels and prevent atherogenesis. Natural dietary compounds have been studied for their beneficial health effects for decades. Diet and nutritional interventions based on the use of natural bioactive compounds is an effective strategy for remodeling gut microbiota composition and improving human health. This review focuses on the mechanisms by which TMAO promote atherosclerosis, the microbes that contribute to TMA formation, the enzymes involved, and the potential of natural dietary compounds that contribute to TMAO reduction and attenuate TMAO-induced atherosclerosis.
Keywords: Cardiovascular disease; TMAO; Gut microbiota; Natural dietary compounds
| 1. Introduction | ▴Top |
1.1. Biosynthesis of trimethylamine-N-oxide (TMAO)
There is growing recognition of the role of diet in modulating the composition and metabolic activity of the human gut microbiota (Conlon and Bird 2015). Trimethylamine (TMA)-containing compounds are sources of TMAO. Phosphatidylcholine (also known as lecithin), choline, and betaine, which are abundant in red meat, egg and milk, participate in intestinal microorganism metabolism and are responsible for converting to TMA (Wang et al. 2011; Wang et al. 2014). L-Carnitine, found predominantly in red meat, is subsequently identified as an alternative precursor of TMA (Koeth et al. 2013). L-Carnitine, an important nutrient for human health, is a quaternary amine compound (Meadows and Wargo 2015). L-Carnitine transports fatty acids into the mitochondria, particularly in cardiomyocytes, thereby facilitating fatty acid breakdown (β-oxidation) and energy derivation (Pekala et al. 2011). 25% L-carnitine can be acquired through endogenous biosynthesis from lysine in the liver, kidney and brain (Flanagan et al. 2010) but our daily demand is largely met through dietary intake from carnitine-containing food, mainly from animal sources such as red meat and dairy products as there is limited carnitine availability in plants (Meadows and Wargo 2015). The bioavailability of L-carnitine varies due to dietary composition. Bioavailability of L-carnitine in vegetarians who are adapted to low-carnitine diets is higher (66–86%) than regular red-meat eaters who are adapted to high-carnitine diets (54–72%) (Flanagan et al. 2010; Rebouche and Chenard 1991). The environment of gut lumen is a complex microbial ecosystem. Intestinal microbiota has been reported to participate in the global metabolism of their host (Dore et al. 2013). They play a role in immunity and defense, digestion and metabolism, inflammation and cell proliferation (Dore et al. 2013). Recent research has focused on gut microbiota-generated metabolites for their impact on regulating host physiology and disease development (Lee and Hase 2014). TMAO is an intestinal microbiota-dependent metabolite. Plasma levels of TMAO are markedly suppressed after the administration of antibiotics and then reappear after withdrawal of antibiotics, suggesting that gut microbiota is required to form TMAO (Tang et al. 2013; Wang et al. 2011). As there are no animal enzymes to break down carnitine, unabsorbed L-carnitine is mostly metabolized by microorganisms in the large intestine to TMA (Flanagan et al. 2010). Additionally, Koeth et al. (2014) suggested another sequential microbial reaction of L-carnitine metabolism (Koeth et al. 2014). Their study showed that following L-carnitine administration, γ-butyrobetaine (γ-BB) is produced as an intermediate metabolite by gut microbiota and then into TMA (Koeth et al. 2014). TMA is subsequently metabolized by flavin monooxygenases (FMOs), a liver phase I enzyme, to TMAO (Liu et al. 2015) (Figure 1). Marine fish is another notable dietary source of TMAO (He and Chen 2018). TMAO serves as an osmoregulatory compound in marine fish, particularly gadoids, and serves as a source of TMA and dimethylamine (DMA) formation in such fish after harvest (Parkin and Hultin 1982).
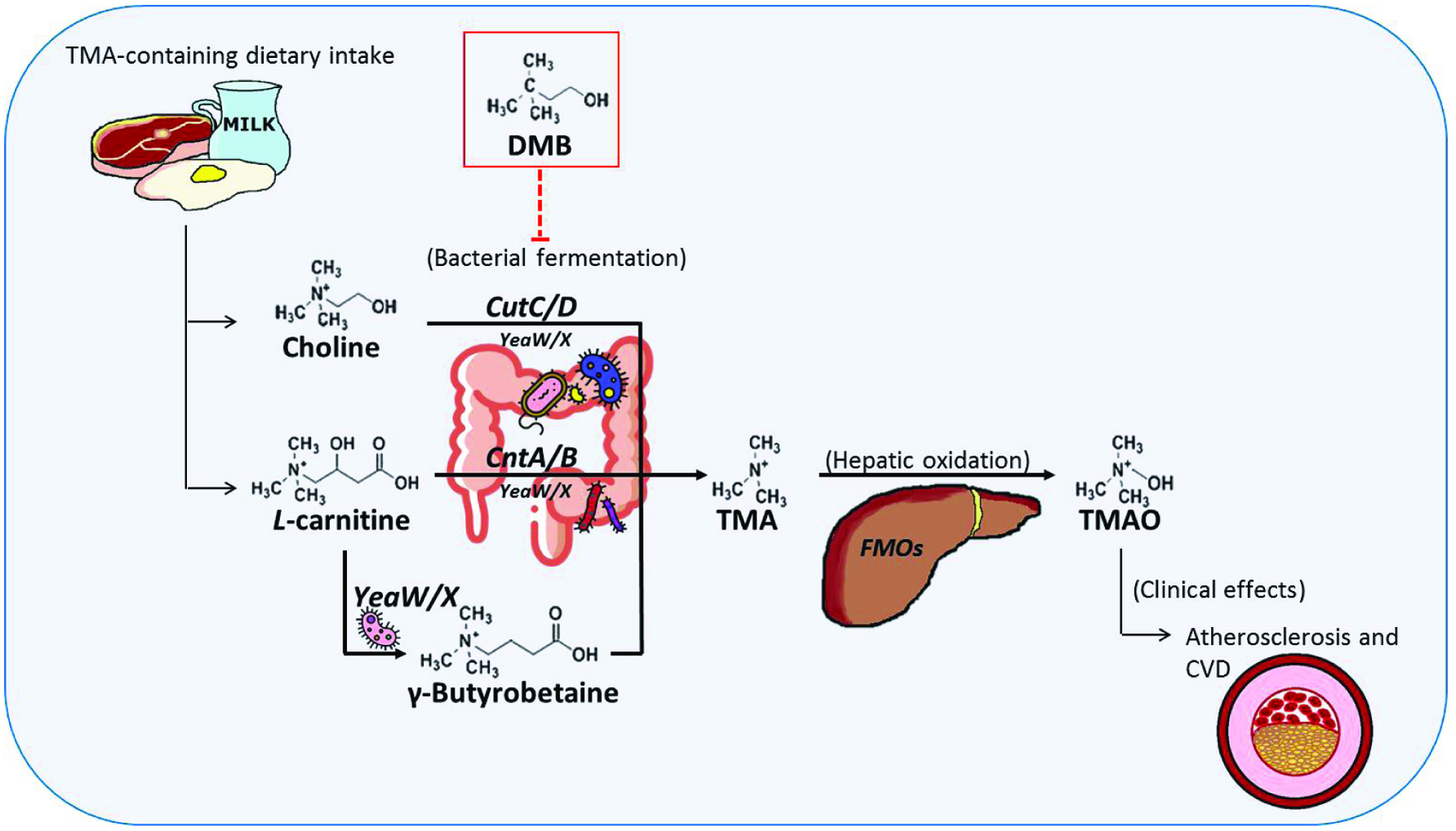 Click for large image | Figure 1. Biosynthesis of gut microbiota-dependent metabolites and atherosclerosis and the effects of DMB. |
1.2. The enzymes contribute to the precursor-TMAO pathway
1.2.1. Glycyl radical enzyme complex—choline-TMA lyase (CutC/D)
Choline is a quaternary amine compound with a trimethylammonium moiety which serves as one of the precursors for TMA. As study by Zeisel et al. (1983) found that humans and rats ingesting choline-containing compounds excrete TMA in their urine (Zeisel et al. 1983). Hayward and Stadtman (1959), using carbon balance of choline fermentation by Vibrio cholinicus (closely related to Desulfovibrio desulfuricans), suggested that the fermentation product was TMA (Hayward and Stadtman 1959). Despite studies on this microbial metabolic pathway (Asatoor and Simenhoff 1965; De La Huerga et al. 1951), nothing was known about the enzyme responsible for choline utilization and the underlying genetics or biochemical mechanisms until 2012. Craciun and Balskus (2012) identified a gene cluster responsible for anaerobic choline degradation and TMA formation in Desulfovibrio desulfuricans American Type Culture Collection (ATCC) 27774, using bioinformatics analysis and electron paramagnetic resonance spectroscopy (Craciun and Balskus 2012). This sulfate-reducing bacterium had previously been reported to form TMA from choline (Baker et al. 1962; Hayward and Stadtman 1959). A glycyl radical enzyme is predicted to promote the carbon (C)-nitrogen (N) bond cleavage for TMA generation, named the genome of this portion the choline utilization (cut) gene cluster (Craciun and Balskus 2012). Using gene knockout and heterologous expression approaches, Craciun and Balskus (2012) confirmed the cut gene cluster is involved in choline utilization. From phylogenetic analysis, this choline-TMA lyase CutC and its activator CutD possess a biochemical function distinct from other glycyl radical enzymes (Craciun and Balskus 2012). Besides, these two enzymes (CutC/D) are responsible for the initial C–N bond cleavage in choline degradation, expression of either enzyme on its own completely abolished TMA production (Craciun and Balskus 2012). Site-directed mutations of either Gly821 or Cys489 residues prevented TMA formation, demonstrated the essentiality of these conserved active site residuals to involve in radical catalysis and for glycyl radical enzyme function (Craciun and Balskus 2012; Lehtio and Goldman 2004).
1.2.2. A two-component carnitine Rieske-type oxygenase/reductase (CntA/B)
Although we know that gut microbiota are responsible for TMAO formation from carnitine, the underpinning genetic and biochemical mechanisms remain unclear. In 2014, a publication in the Proceedings of the National Academy of Sciences of the United States of America (PNAS) using bioinformatics approaches, first revealed the enzyme involved in carnitine metabolism as being a two-component Rieske-type oxygenase/reductase, designated as CntA/B (Zhu et al. 2014). CntA belongs to a group of uncharacterized Rieske-type proteins and has an unusual “bridging” glutamate instead of aspartate residue, which plays a role in electron transfer between the Rieske center and the catalytic mononuclear iron center, and might carry out oxidative reaction. CntB predicted to be a NAD(P)-dependent reductase, consists of a FAD-binding domain and a plant-type ferredoxin (Zhu et al. 2014). Using Acinetobacter baumannii ATCC 19606 as the model, the authors demonstrated that cntAB is essential and sufficient in carnitine degradation to TMA, known as carnitine-TMA lyase (Zhu et al. 2014). These enzymes are supposed to cleave C–N bond of carnitine to produce TMA and a four-carbon (C4) skeleton, which subsequently enters the tricarboxylic acid cycle utilize as a carbon source in the intact cells of Acinetobacter calcoaceticus (Seim et al. 1982; Zhu et al. 2014). The presence of cntAB was predominantly related to γ-Proteobacteria (Rath et al. 2017). In addition, Koeth et al. (2013) demonstrated that enriched proportions of the genus Prevotella exhibit higher plasma TMAO levels after L-carnitine ingestion (Koeth et al. 2013).
1.2.3. An additional enzyme complex shows carnitine-TMA lyase activity (YeaW/X)
The γ-BB is thought to be an intermediate in gut microbe-dependent formation of TMAO from L-carnitine (Koeth et al. 2014; Rebouche et al. 1984). Besides, L-carnitine degradation to γ-BB is initialized in the mouse ileum, anatomically proximal to those for TMA production which appears restricted to the bacteria-rich cecum and the colon (Koeth et al. 2014). Koeth et al. (2014) searched for unknown microbial enzymes in gene clusters within a reference genome covering enzymes for malate or succinate synthesis and a presumed betaine-carnitine-choline transporter, allowed the authors to identify YeaW/X as a new microbial enzyme complex for TMA generation (Koeth et al. 2014). Interestingly, recombinant microbial YeaW/X complex showed up it can metabolize multiple TMA precursors (γ-BB, L-carnitine, choline, and betaine) (Koeth et al. 2014) (Figure 1). The study also found that plasma TMAO levels were significantly associated with the relative abundances of Akkermansia, within the cecum recovered from the γBB supplemented mice (Koeth et al. 2014). In contrast, an in vitro study demonstrated that Akkermansia does not convert choline to TMA (Romano et al. 2015). YeaW/X shows close sequence similarity to CntA/B and was then proposed to be the key component of the third TMA synthesis pathway (Rath et al. 2017).
1.2.4. Flavin monooxygenases (FMOs)
Flavin monooxygenases (FMOs) are microsomal phase I enzymes that incorporate an atom of molecular oxygen into a wide variety of nitrogen- and sulfur-containing drugs and xenobiotics (Krueger and Williams 2005). There are many FMO isoforms, however FMO1, FMO2 and FMO3 are the major drug and xenobiotic metabolizing isoforms and are expressed in a tissue-, species-, sex-, and developmental-specific manner (Siddens et al. 2008). FMOs activity in hepatic tissue homogenates indicated significantly higher enzymatic activity in female than in male mice (Bennett et al. 2013; Romano et al. 2015; Wang et al. 2011). In the liver, FMO3 is responsible for converting TMA to TMAO, which exhibits 10-fold higher specific activity than FMO1 (Bennett et al. 2013). While individuals with mutations in FMO3 would cause trimethylaminuria, as they have diminished capacity to oxidize the dietary-derived fish-like odor amine TMA to its odorless metabolite TMAO and thus excrete relatively large amounts of TMA in their urine, sweat and breathe and often described as having “fish odor syndrome” (Treacy et al. 1998).
1.3. TMA-producing microbiota
The gut microbiota is a flourishing field with many novel findings that provide a new perspective towards the development and progression of diseases (Li and Tang 2017). Currently, information on the composition of TMA-producing microbiota in the intestine is scarce due to their low abundance. However, even low levels of colonization of TMA-producing bacteria are sufficient for TMA synthesis and TMAO accumulation (Romano et al. 2015). Some of the metabolic pathways that generate TMA have only recently been elucidated, 16S ribosomal RNA (rRNA) gene sequencing analysis is unavoidably the main way to correlate microbiome composition to plasma TMAO levels (Falony et al. 2015). However, Rath et al. (2017) established a functional assay for the key genes of the major TMA-forming pathways and revealed that 16S rRNA gene sequence studies cannot adequately predict the TMA-producing potential (Rath et al. 2017).
Choline utilization showed uneven distribution between common phyla found in the human gut (Craciun and Balskus 2012). Although conversion potential can be detected in Firmicutes, Proteobacteria, and Actinobacteria, they are completely absent from Bacteroidetes (Craciun and Balskus 2012; Martinez-del Campo et al. 2015). Campo et al. (2015) also observed a discontinuous distribution of the cut gene cluster across most microbial species (Martinez-del Campo et al. 2015). Desulfosporosinus and Proteus are the only two bacterial genera that universally harbor the cut gene cluster (Martinez-del Campo et al. 2015). Putative genes encoding CntA/B were mainly observed in Proteobacteria, especially γ-Proteobacteria (mostly derived from Escherichia and Acinetobacter) and a few β-Proteobacteria, ɛ-Proteobacteria and Firmicutes (Rath et al. 2017; Zhu et al. 2014). YeaW/X has been described in several taxa from β-Proteobacteria as well as from a few Firmicutes (Koeth et al. 2014; Zhu et al. 2014) (Table 1).
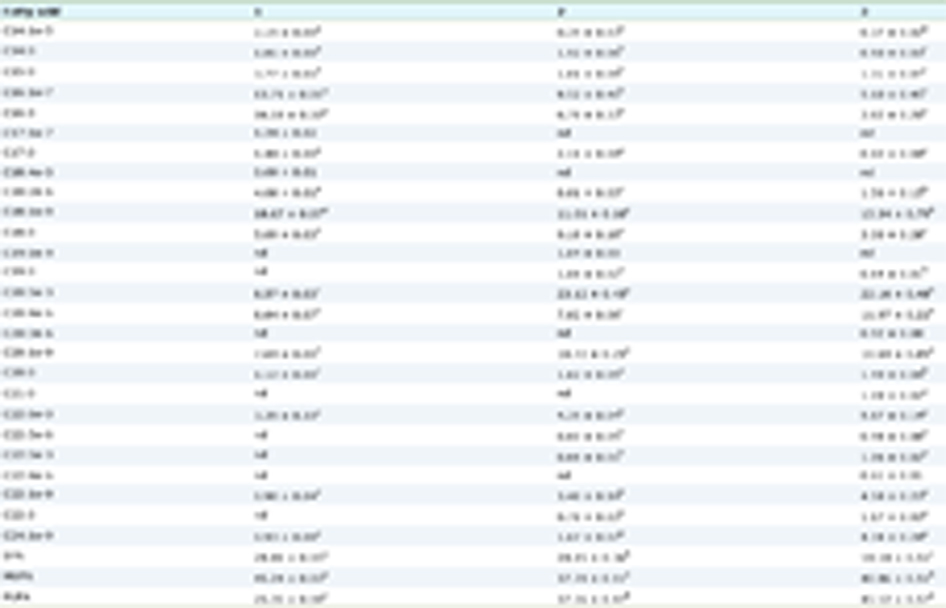 Click to view | Table 1. Three key TMA-generating enzymes and the corresponding TMA producers |
| 2. TMAO and atherosclerosis | ▴Top |
TMAO has recently been discovered as an important intestinal microbiota-dependent metabolite that causes cardiovascular disease (CVD), particularly atherosclerosis (Bennett et al. 2013; Liu et al. 2015; Tang et al. 2013; Wang et al. 2011). It has been shown that increasing levels of TMAO was dose-dependently associated with the presence of CVD (Wang et al. 2011). Tang et al. (2013) reported an association between the history of CVD and elevated fasting plasma levels of TMAO (Tang et al. 2013). In atherosclerosis-prone mice (C57BL/6J ApoE−/− mice), normal chow diet supplemented with high amounts of choline (1.0%) or TMAO (0.12%) increased total aortic root atherosclerotic plaque area (Wang et al. 2011). However, in a large human clinical cohort study, plasma carnitine levels in subjects (n = 2,595) were independently associated with major adverse cardiac events (MACE, including death, myocardial infarction, stroke and revascularization) (Koeth et al. 2013). Only among those subjects with concomitant high plasma TMAO levels, plasma levels of carnitine appeared to be associated with CVD risk (Koeth et al. 2013). Additionally, elevated plasma levels of choline and betaine are each associated with incident MACE risk independent of traditional risk factors (Wang et al. 2014). High choline and betaine levels were only associated with higher risk of future MACE with concurrent increased levels of TMAO (Wang et al. 2014). These results are consistent with intestinal microbiota-dependent metabolite TMAO, but not dietary precursors, serving as the culprit of enhancing atherogenesis manner (Koeth et al. 2013; Wang et al. 2014). CVD has become a ubiquitous cause of morbidity and a leading contributor to mortality in most countries. Atherosclerosis, a major component of CVD, has properly been considered a public health problem of industrialized countries, perhaps reflecting greater exposure to certain risk factors with rising standards of living (O’Connor et al. 2001). Thus, the need for public health measures to limit its impact is expanding worldwide.
Atherosclerosis is an arterial disease. The process of atherogenesis is largely caused by the accumulation of lipids within the artery wall and buildup as a plaque. The lesions of atherosclerosis represent a series of highly specific cellular and molecular responses that can be described as an inflammatory disease (Ross 1999). Additonally, the inflammation is present in the vessel wall throughout all stages of the disease until the final pathophysiologic steps (Koenig and Khuseyinova 2007). The earliest type of lesion of atherogenesis is called fatty streak, a pure inflammatory lesion, consisting of arterial intimal accumulations of foam cells (lipid-laden macrophages) and T lymphocytes intermixed with smooth muscle cells. Gradually, the influx of these cells is preceded by the extracellular deposition of amorphous and membranous lipids, in persons with high plasma cholesterol (O’Connor et al. 2001; Ross 1999). Plaque buildups cause the inside of the arteries to narrow over time, which block the blood flow. The endothelium normally forms a barrier to the passage of blood constituents into the artery wall. Recent hypothesis emphasizes endothelial dysfunction, instead of denudation, is the first step in atherosclerosis (Ross 1999). Possible causes of endothelial dysfunction leading to atherosclerosis include elevated and modified LDL; free radicals, hypertension, and diabetes; infectious microorganisms such as Chlamydia pneumonia (Ross 1999). Host usually elicits proinflammatory cytokines and stimulates increased expression of cellular adhesion molecules which enhancing leukocyte adhesion, to defend extravascular infections (O’Connor et al. 2001). Oxidized lipids, such as oxidized LDL (oxLDL), contribute to recruitment of inflammatory cells and their activation in atherosclerosis (Shah 2003), placing oxLDL in a central role for the development of atherosclerosis. OxLDL has a large number of biological actions and consequences, including injuring endothelial cells, expressing adhesion molecules, recruiting leukocytes and the formation of foam cells (Jessup et al. 2004). Inflammatory cells are probably recruited into the atherosclerotic plaques by adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1), and chemokines, such as monocyte chemoattractant protein-1 (MCP-1). They are then activated in the vessel wall. MCP-1 is one of the most important chemokines that regulates migration and infiltration of macrophages (Koenig and Khuseyinova 2007). Besides, cell adhesion molecules on endothelial cells such as intercellular adhesion molecules-1 (ICAM-1), VCAM-1, E-selectin, and P-selectin also promote leukocyte migration (Adams and Shaw 1994). Proinflammatory cytokines such as interleukin-6 (IL-6) stimulates production of interleukin-1β (IL-1β), tissue necrosis factor-α (TNF-α) and transforming growth factor-β (TGF-β) (Huber et al. 1999).
In LDLR−/− mice, TMAO injection activates inflammatory gene expression of cyclooxygenase-2, IL-6, E-selectin, and ICAM-1 in aortas (Seldin et al. 2016). Besides, suggesting that TMAO exerts inflammatory effects on human aortic endothelial cells through activation of signaling of mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NF-κB) (Seldin et al. 2016). One percent choline supplemented diet showed markedly enhanced lipid-laden macrophage development (Wang et al. 2011). TMAO promoted cholesterol accumulation within macrophages, by inducing scavenger receptors such as CD36 and SR-A1 linked to atherosclerosis, and foam cell formation in ApoE−/− mice (Wang et al. 2011). Theoretically, TMAO may cause lipid accumulation by facilitating lipid influx, enhancing lipid synthesis, and suppressing lipid efflux and lipid clean-up (Liu et al. 2015). TMAO inhibits cholesterol efflux by suppressing reverse cholesterol transport (RCT), a process of HDL to transport cholesterol out of the arterial wall for eliminating atherosclerotic plaques, as well as by blocking bile acid secretion (Koeth et al. 2013; Liu et al. 2015). RCT was also suppressed in mice receiving either choline or carnitine supplementation (Koeth et al. 2013). However, following antibiotic administration, the suppression of RCT was reversed; therefore, TMAO is produced by gut microbes and inhibits RCT (Koeth et al. 2013). Moreover, TMAO disrupting bile acid metabolism through reduced the levels of bile acid synthetic enzymes (CYP7A1 and CYP27A1), bile acid transporters (OATP1, OATP4, MRP2, and NTCP) in the liver and intestinal cholesterol transporters (NPC1L1 and ABCG5/8) (Koeth et al. 2013). As the bile acid pathway is of central importance in cholesterol elimination, blocking this pathway may possibly accelerated atherosclerosis (Figure 2).
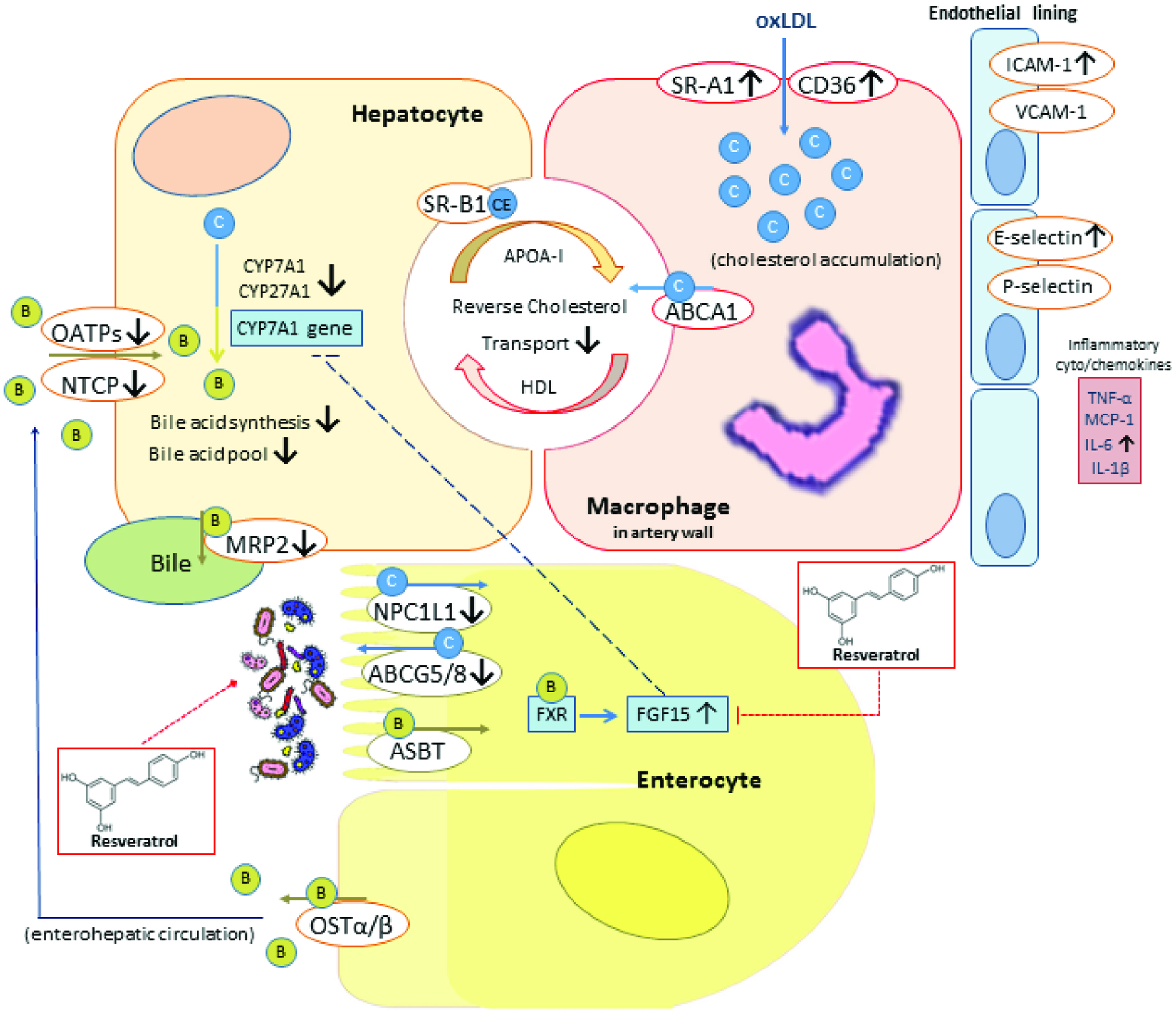 Click for large image | Figure 2. Schematic representation of pro-atherosclerotic ability of TMAO and the effects of resveratrol. |
| 3. Natural compounds in TMAO research | ▴Top |
As TMAO generation is dependent on gut microbiota, gut microbiota has been put forward as a key player in TMAO-induced pathogenesis. Administration of antibiotics can successfully block the pathway of TMA formation governed by gut microbiota and attenuated TMAO-promoted severity of atherosclerosis (Koeth et al. 2013). However, there are many possible and undesirable side effects of antibiotics and the considerations of antibiotic resistance in bacteria. Diet and nutritional interventions based on the use of natural bioactive compounds may offer one of the possibilities for remodeling gut microbiota composition and improving human health (Banikazemi et al. 2018; He and Chen 2018).
3.1. Resveratrol
Natural polyphenolic compounds exhibit many beneficial health effects, such as anti-oxidation, anti-inflammatory and anti-carcinogenesis. The poor bioavailability of phenolic compounds may possibly be the reason this class of compounds developing into therapeutic agents through modulating structure of gut microbiota (Cardona et al. 2013). Among polyphenols, resveratrol has been demonstrated to improve gut microbiota dysbiosis (Qiao et al. 2014). Resveratrol exerts anti-obesity effects through regulating gut microbiota composition by inducing a decrease in Firmicutes/Bacteroidetes ratio and increasing the growth of Lactobacillus, Bifidobacterium and Akkermansia in mouse model (Qiao et al. 2014). Chen et al. (2016) using resveratrol, also demonstrated its gut microbiota-remodeling activity (decreased Firmicutes/Bacteroidetes ratio and increased the growth of Lactobacillus, Bifidobacterium and Akkermansia) and resulted in TMAO-induced atherosclerosis attenuation in choline-fed ApoE−/− mice (Chen et al. 2016). As mentioned above, TMAO can induce atherosclerosis through affecting bile acid metabolism by inhibiting bile acid synthetic enzymes (CYP7A1 and CYP27A1) in the liver (Koeth et al. 2013). Chen et al. (2016) observed an increase in CYP7A1 mRNA and protein expression, which enhanced hepatic bile acid neosynthesis in the resveratrol-treated mice. This observation was partially mediated through the enterohepatic FXR-FGF15 axis (Figure 2), which mediated by bile acid, and via gut microbiota remodeling to one which has been associated with a better metabolic profile (Chen et al. 2016). Resveratrol also enhanced bile acid deconjugation and fecal excretion (Chen et al. 2016).
3.2. 3,3-Dimethyl-1-butanol (DMB)
As mentioned above, Craciun and Balskus (2012) identified a choline-TMA lyase complex (CutC/D). Accordingly, we can utilize our understanding of key microbial genes in an attempt to “drug the bacteria”. Wang et al. (2015) cloned and expressed the cutC/D genes as a microbial choline-TMA lyase activity system to survey natural or synthetic structural analogues of choline for their effects on CutC/D activity. One of the natural analogues examined, 3,3-dimethyl-1-butanol (DMB), showed choline-TMA lyase inhibitory activity (Wang et al. 2015). DMB is an analogue of choline, where the quaternary amine nitrogen of choline is replaced with a carbon, found as a natural product in some cold-pressed extra virgin olive oil, grape seed oil and red wine (Wang et al. 2015). It is important to note that DMB is relatively non-toxic and it is not an antibiotic, which makes it less likely for resistance to develop. In a further study, DMB was shown to non-lethally inhibit TMA formation from cultured microbes and also inhibits TMA formation from multiple substrates (e.g. choline, carnitine, γ-BB, phosphatidylcholine) in polymicrobial cultures (Wang et al. 2015). However, DMB is shown to inhibit distinct microbial TMA lyases. DMB failed to inhibit CntA/B-catalyzed cleavage of carnitine to TMA (Wang et al. 2015). Despite the homology to CntA/B, YeaW/X cleavage activity is inhibited by DMB for conversion of carnitine into TMA (Wang et al. 2015). In ApoE−/− mice, DMB significantly inhibited TMAO, diet-enhanced macrophage foam cell formation and aortic root lesion development in both male and female mice fed with choline diet compared to diet without DMB addition (Wang et al. 2015). This observation corresponds with previous study of the author for no TMAO increase and no increased cholesterol in macrophages in mice supplemented with DMB (Wang et al. 2011). Interestingly, DMB decreased Clostridiales, which has been reported as a TMA-producer (Craciun and Balskus 2012; Martinez-del Campo et al. 2015) (Table 1) and is associated with atherosclerotic lesion (Koeth et al. 2013). In contrast, Bacterioidetes was inversely associated with atherosclerotic lesion upon increase in DMB administration (Wang et al. 2015). Therefore, DMB potentially inhibited some microbial TMA lyases and reorganized the gut microbial community (Figure 1).
| 4. Conclusion | ▴Top |
Evidences show that the gut microbial pathway is involved in TMAO generation, a metabolite linked to atherosclerosis. Since antibiotic treatments have side effects and the concern about bacteria resistance, the challenge is to develop new therapeutic strategies. For this purpose, several approaches have emerged such as the use of probiotics (Boutagy et al. 2015; Degirolamo et al. 2014) and change in dietary consumption. Indeed, natural products are considered as a rich source of bioactive molecules. Natural polyphenols as bioactive compounds with many promising beneficial effects on human disease development are reported to regulate gut microbiota composition, hence may serve as new agents against TMAO-enhanced atherosclerosis. Moreover, databases for cutC and cntA sequences are available at http://193.175.244.101/TMA/. We can use molecular docking studies to search for natural small molecules as an inhibitor (Tippani et al. 2013) for distinct microbial TMA lyase which can lead to novel way for non-lethally preventing atherogenesis and regulating gut microbiota community.
This study was supported by the Ministry of Science and Technology (105-2320-B-002-031-MY3, 105-2628-B-002-003-MY3).
Conflict of interest
There are no conflicts to declare.
| References | ▴Top |