| Journal of Food Bioactives, ISSN 2637-8752 print, 2637-8779 online |
| Journal website www.isnff-jfb.com |
Mini-Review
Volume 13, March 2021, pages 12-19
Utility of animal models of Alzheimer’s disease in food bioactive research
Klaus W. Lange
Department of Experimental Psychology, Institute of Psychology, University of Regensburg, 93040 Regensburg, Germany. Tel: +49 941 9433815; Fax: +49 941 9434496; E-mail: klaus.lange@ur.de
DOI: 10.31665/JFB.2021.13255
Received: March 16, 2021
Revised received & accepted: March 28, 2021
| Abstract | ▴Top |
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by globally impaired cognition. AD research in animals has shown a substantial deficit in translational relevance. The most extensively used transgenic mouse models overexpress human genes associated with rare familial early-onset AD, which results in the formation of amyloid plaques. However, the most common form of AD (late-onset sporadic AD) is a multifactorial disorder involving different cytotoxic factors, including neurofibrillary pathology. Transgenic mice studies have been valuable in elucidating pathogenetic mechanisms that may be relevant to human AD. However, their utility in the development of novel treatment strategies and as preclinical testbeds of new drugs has been unsatisfactory. Animal models have so far failed to demonstrate predictive value in regard to novel therapies of AD, including the use of bioactive food components. While many therapeutic approaches assessed in animals have shown promising results, attempts to translate these findings to people with AD have been disappointing. Food scientists should be aware that the available animal models appear to be unable to predict clinical success in humans. Therefore, food bioactive research should focus on human-centric preventive approaches to AD in clinically meaningful settings rather than on highly questionable preclinical research in animals.
Keywords: Alzheimer’s disease; Animal models; Food bioactives; Prevention; Treatment
| 1. Introduction | ▴Top |
Alzheimer’s disease (AD) is characterized clinically by a progressive, global impairment of cognitive abilities, including memory, learning, perception, attention, planning, decision-making and language (Lange et al., 1995; Scheltens et al., 2016; Weintraub et al., 2012). The disorder is associated with a marked decline in activities of daily life, with symptoms normally appearing late in life following a long prodromal period of brain pathology over several decades. Given its increasing global prevalence, AD has become a major public health challenge worldwide (Prince et al., 2015). AD is a proteinopathy associated with the aggregation and accumulation of misfolded proteins (Kosik and Shimura, 2005; DeTure and Dickson, 2019). The neuropathological hallmarks of the progressive neurodegeneration in AD, needed to make a definite diagnosis of the disorder, are (1) extracellular amyloid deposits (senile plaques) containing misfolded amyloid-β (Aβ) peptides and (2) intracellular aggregates of hyperphosphorylated tau protein forms (neurofibrillary tangles) (Butterfield and Lauderback, 2002; Crews and Masliah, 2010; DeTure and Dickson, 2019; Price et al., 1991). It is noteworthy, however, that amyloid plaques and neurofibrillary tangles are commonly found in the brains of elderly people, although usually to a lesser degree in those with unimpaired cognition. These changes constitute evidence of a biological process and not necessarily a state of disease (Davis et al., 1999). Thus, neither the clinical symptoms nor the neuropathological alterations alone can establish the diagnosis of AD, indicating the limited diagnostic specificity of the clinical and pathological criteria. Major pathophysiological mechanisms in the complex pathogenesis of AD include oxidative stress and chronic neuroinflammation. The aggregation of Aβ can lead to the generation of large quantities of free radicals, such as active oxygen and nitrogen species, causing oxidative stress and thereby accelerating neuronal dysfunction and ultimately leading to widespread neuronal death and brain atrophy (Butterfield and Lauderback, 2002; Querfurth and LaFerla, 2010).
The majority of cases of AD are late-onset sporadic forms of unknown etiology, without any obvious familial aggregation. However, familial studies have determined an autosomal dominant mode of inheritance in rare forms of AD, with systematic linkage analyses characterizing APP, PSEN1 and PSEN2, coding for amyloid precursor protein, presenilin-1 and presenilin-2, respectively, as the causative genes for early-onset (<60 years) forms of AD (Cacace et al., 2016). The rare mutations in these genes have been shown to selectively enhance the production of Aβ (Scheuner et al., 1996). These mutations have given rise to the amyloid cascade hypothesis of AD, with Aβ imbalance and toxicity thought to be the main causes of synaptic dysfunction and neurodegeneration (Golde, 2003; Hardy and Higgins, 1992; Karran et al., 2011; Selkoe, 2001). Furthermore, epidemiological studies have shown that 40–50% of individuals with AD are carriers of the ε4-allele of the apolipoprotein E gene (ApoE), compared to 15% in healthy people (Genin et al., 2011; Strittmatter et al., 1993). The presence of ApoE4 appears to have no causal links to AD and to be an allele-specific risk factor of late-onset AD. The APP, PSEN1, PSEN2, and ApoE-ε4 genes have been estimated to account for 30–50% of genetic variance in AD (Tanzi, 2012; Tanzi and Bertram, 2001).
The symptomatic pharmacotherapy of AD, including the administration of acetylcholinesterase inhibitors and N-methyl-D-aspartate receptor antagonists, has shown no significant efficacy in AD (Scheltens et al., 2016). The success of new drugs in clinical trials has been low, with most failing to show efficacy in randomized controlled trials (Cummings et al., 2014). Furthermore, no disease-modifying interventions capable of retarding the progressive neurodegenerative process are currently available (Scheltens et al., 2016). The prevention of AD is therefore a major public health challenge (Lange, 2018a) requiring the development of novel interventions capable of delaying the onset of the disease. Various lifestyle factors, such as obesity, diabetes, hypertension, “unhealthy” diet and smoking may increase the risk of AD (Lange, 2018a; Sohn, 2018; Yu et al., 2020). The elimination of modifiable lifestyle factors has been estimated to decrease the incidence of dementia by at least 30% (Bruijn et al., 2015), highlighting the potential value of modifications in lifestyle-related factors in the primary prevention of AD (Norton et al., 2014; Wu et al., 2016). Diet, in particular, is receiving increasing recognition as an important modifiable factor, since it can affect alterations of brain and behavior related to aging and disease (Gustafson et al., 2015; Huhn et al., 2015). Dietary approaches to AD include the Mediterranean diet (Gu et al., 2010; Lange, 2019), ketogenic diets (Lange et al., 2017) and some medical foods (Lange et al., 2019a; Lange et al., 2019b). The potential therapeutic benefits of various food bioactives, such as natural antioxidants, probiotics and polyunsaturated fatty acids, on AD have been investigated (Lange, 2018b, 2020a, 2020b; Lange and Li, 2018; Lange et al., 2020). Natural polyphenols, such as flavonoids, appear to have the potential to modulate the neuropathological alterations and cognitive impairment associated with AD through various mechanisms, including the modulation of oxidation and inflammation as well as of Aβ metabolism, catabolism and oligomerization (Gaudreault and Mousseau, 2019). Furthermore, prospective studies in humans have found an association between high levels of dietary flavonoid consumption and a lower risk of AD (Commenges et al., 2000; Engelhart et al., 2002; Holland et al., 2020).
| 2. Animal models of AD | ▴Top |
As in other diseases, properly conducted animal models may be useful in the search for preventive measures or a cure for AD. The development of animal models with cognitive profiles and pathological changes similar to those seen in humans with AD is the goal of translational research (Sharbaugh et al., 2003). Such models could be used in preclinical studies assessing the potential of novel therapeutics. Although the “amyloid hypothesis” of AD (Hardy and Higgins, 1992; Mattson, 2004) has been questioned (Schnabel, 2011), it nevertheless forms the basis of a wide range of animal models mimicking the human disease. Various animal models have been used as a research tool in the investigation of the pathogenesis and pathophysiology of AD as well as the preclinical development of novel therapeutics. Major models are mice transgenically overexpressing mutated human genes that lead to the formation of amyloid plaques (Esquerda-Canals et al., 2017; Myers and McGonigle, 2019). Less commonly used animal models include zebrafish (Newman et al., 2014) and invertebrates, such as Drosophila melanogaster and Caenorhabditis elegans (Bouleau and Tricoire, 2015; Hannan et al., 2016; Tsuda and Lim, 2018). Transgenic technology allows the reproduction of the cause of the rare familial form of AD by transfecting mutant human APP. Transgenic mice carrying gene mutations associated with familial AD have been found to express high brain levels of mutant APP and to develop Aβ deposits structurally similar to those seen in humans (Games et al., 1995), with mice carrying multiple gene mutations accumulating Aβ faster than single-mutation mice (Janus and Welzl, 2016). Aged non-human primates may develop structural and neurochemical changes similar to AD (Gearing et al., 1994; Gearing et al., 1997; Perez et al., 2016; Picq et al., 2012). However, their use in research poses numerous practical and ethical challenges.
2.1. Food bioactives in animal and human studies of AD
Natural antioxidants, such as the polyphenols resveratrol and (-)epigallocatechin-3-gallate (EGCG), have been proposed as potential treatment options in AD. Plant polyphenolics are thought to have various beneficial health effects due to their antioxidant activity (Urquiaga and Leighton, 2000). The stilbenoid resveratrol (trans-3,5,4′-trihydroxystilbene) from edible plants, such as grapes, berries and grains, has been shown to possess neuroprotective properties in various experimental settings and to reduce toxic Aβ aggregation (Lange and Li, 2018; Richard et al., 2011; Smid et al., 2012). For example, in an APP/PSEN1 transgenic mouse model of AD, the long-term administration of resveratrol over 10 months markedly reduced memory loss and the amyloid burden in the brain (Porquet et al., 2014). The effects of resveratrol on Aβ1–42-induced impairment of learning and memory have been examined in mice following microinfusion of Aβ1–42 bilaterally into hippocampal CA1 subregions (Wang et al., 2016). In this study, resveratrol reversed Aβ-induced memory impairment in the Morris water maze task (Wang et al., 2016). In another study, resveratrol was shown to protect rats from Aβ-induced hippocampal neuron loss and memory impairment (Huang et al., 2011). The effects of resveratrol supplementation have also been investigated in individuals with age-related cognitive decline. In a small sample of people with mild cognitive impairment, the supplementation with resveratrol (200 mg daily) over 26 weeks preserved hippocampal volume and improved resting-state functional connectivity of the hippocampus in comparison with controls (Köbe et al., 2017). Whether these benefits of resveratrol supplementation can be extended to cognitive functioning is unknown. Chronic administration of resveratrol for 52 weeks in people with mild to moderate AD (500 mg orally once daily; dose escalation by 500-mg increments every 13 weeks; final dose of 1,000 mg twice daily) showed no effects on Aβ42 levels or tau/phospho-tau 181 in plasma or cerebrospinal fluid, hippocampal volume or entorhinal cortex thickness (Turner et al., 2015). However, the administration of resveratrol increased brain volume loss compared to placebo (Turner et al., 2015). These findings suggest no beneficial effects of long-term resveratrol supplementation.
The poor bioavailability of resveratrol is a limiting factor in its supplementation. The naturally dimethylated resveratrol analog pterostilbene (trans-3,5-dimethoxy-4′-hydroxystilbene), contained in blueberries, may have greater therapeutic potential in AD (Lange and Li, 2018) due to its more lipophilic nature (Kapetanovic et al., 2011). For example, oral administration of pterostilbene has shown better efficacy than resveratrol in improving cellular oxidative stress and cognitive impairment in an AD mouse model (Chang et al., 2012). However, findings from human trials are not available.
Bioactive components contained in tea have been proposed to be of value in the prevention of neurodegeneration (Chen et al., 2018). Several lines of evidence suggest that tea consumption may decrease the incidence of AD (Mandel et al., 2008). In particular, the polyphenolic flavonoid EGCG appears to show neuroprotective activity in preclinical models. In transgenic mice with an overexpression of Aβ, the administration of EGCG has been found to significantly decrease Aβ deposition and oxidative stress, to modulate tau pathology and to reduce cognitive impairment, particularly in spatial memory tasks (Rezai-Zadeh et al., 2005; Rezai-Zadeh et al., 2008). The preclinical findings of neuroprotective effects of EGCG in AD models have recently been reviewed (Youn et al., 2021). Due to its high levels of EGCG (Lin et al., 2003), the efficacy of green tea in age-related cognitive decline in humans has been examined in several randomized controlled trials. A small randomized, double-blind, placebo-controlled study found beneficial effects of combined green tea extract and L-theanine on cognition in individuals with mild cognitive impairment (Park et al., 2011). In contrast, another 12-month double-blind, randomized controlled study examining the effects of green tea consumption in aged individuals with cognitive dysfunction found no significant differences in changes in the Mini-Mental State Examination between the administration of green tea powder and placebo (Ide et al., 2016). Limitations of this study include a small sample size, a short follow-up duration and a lack of psychiatric assessment. The clinical evidence regarding EGCG in AD has recently been reviewed (Lange et al., 2021).
Potential effects of omega-3 fatty acids in the prevention of AD are suggested by preclinical findings. For example, the long-term administration of omega-3 PUFAs in animal models for more than 10% of the lifespan has been found to be capable of reducing Aβ and improving cognitive function (Hooijmans et al., 2012). Other animal studies have reported an association between omega-3 PUFA intake and a decrease in Aβ42 blood levels (Green et al., 2007; Lim et al., 2005), neuro-inflammation (Cole et al., 2005; Gil, 2002; Lukiw et al., 2005) and cognitive decline (Gamoh et al., 2001; Hashimoto et al., 2002). A systematic review of seven studies examining supplementation of omega-3 PUFAs in AD reported that most of the included studies found no statistically significant benefits of omega-3 supplementation in comparison with placebo. The beneficial effects that were found were confined to improvements in a few cognitive assessment scales in individuals with very mild AD (Canhada et al., 2018). A Cochrane review of three randomized, placebo-controlled trials of high methodological quality examining omega-3 PUFA supplements in 632 participants with mild to moderate AD over 6, 12 and 18 months found no effects on cognition, everyday functioning, quality of life or mental health (Burckhardt et al., 2016). Furthermore, the administration of omega-3 PUFAs for 26 weeks produced improvements in executive functions, gray matter volume and white matter microstructure in healthy elderly people (Witte et al., 2014). However, other studies found no beneficial effects on cognitive health following the intake of fish oil in healthy elderly individuals (Dangour et al., 2010; van de Rest et al., 2008). A Cochrane review also found no benefits of omega-3 PUFA supplementation on cognition in elderly individuals (Sydenham et al., 2012).
In summary, while the results regarding natural antioxidants and omega-3 fatty acids in animal models of AD have been promising, attempts to translate these findings to individuals with AD have been disappointing (see Table 1).
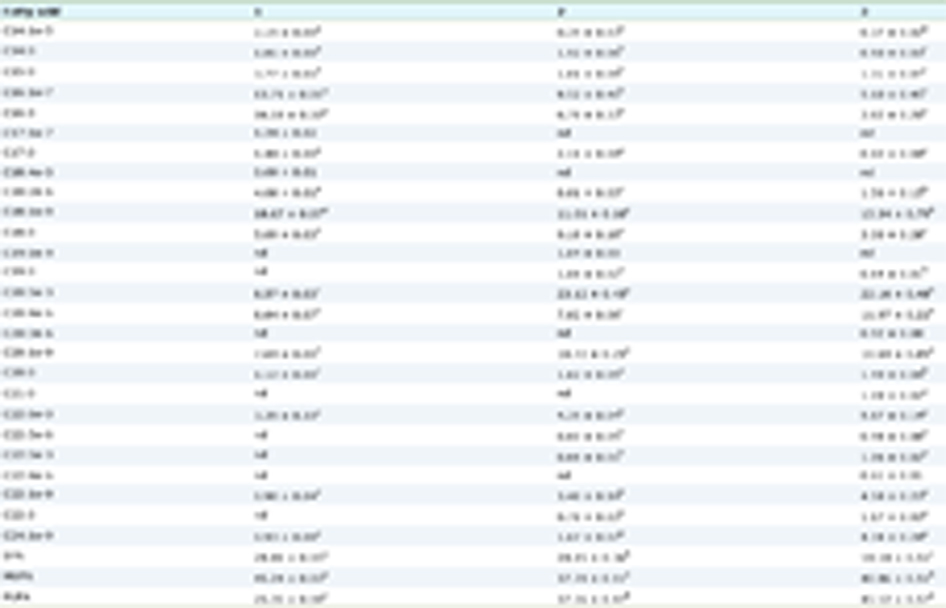 Click to view | Table 1. AD-related effects of natural antioxidants and omega-3 PUFAs in animal models and humans |
2.2. Discrepancy between animal and human studies of AD
The failure of transgenic mice and other animal models to forecast the efficacy of potential anti-AD compounds in humans is not confined to food bioactives (Franco and Cedazo-Minguez, 2014). Reasons why the success of animal models in predicting efficacious treatments in clinical trials has been limited may include qualitative and quantitative differences between models and humans (Laurijssens et al., 2013). For example, an animal model may not truly reflect the human condition, and the target of a novel drug in animals may not be relevant to the disease in humans. In this qualitatively different situation, the key elements of the preclinical model do not play a major role and are involved to different degrees in the causal trajectory in people. A quantitative difference may exist where, although the drug target is relevant, the dosage may be inappropriate due to species differences in pharmacokinetics, target affinity or treatment duration needed.
A major problem related to the currently available AD models is their validation. A perfect animal model of human disease does not exist, since such a model would need to be a scaled down replica of human disease, with all important aspects of cause and pathomechanisms, structural damage and biochemical changes as well as symptoms. Given the complexity of AD, no animal model is ever likely to satisfy these criteria. The criteria commonly used for the evaluation of animal models, i.e. face validity, construct validity and predictive validity, are met to different degrees in Alzheimer models.
Face validity (resemblance of the disease in humans on a phenomenological level, e.g. similar pathology in models and humans) is at least partly satisfied in most animal models. All transgenic mouse models show evidence of cognitive impairment, particularly in tasks assessing spatial memory. However, cognitive impairment is observed much earlier than in AD, with deficits in mice usually occurring coincidently with plaque development, in contrast to several decades following the onset of plaque pathology in humans (Janus and Welzl, 2016). Transgenic mice expressing mutant forms of human proteins related to early-onset AD were developed primarily for their ability to show early cognitive impairment as a therapeutic target. For example, the transgenic Tg2576 mouse, which carries a mutation of human APP, shows amyloid plaques in cortex and hippocampus as well as cognitive deficits at the age of 10–16 months (Frautschy et al., 1998; Hsiao et al., 1996). The development of the phenotype may also be accelerated by mixing two mutations of human APP in the transgene (Mucke et al., 2000) or by crossing the Tg2576 mouse with another animal carrying a human PSEN1 mutation (Holcomb et al., 1998). However, in a number of aspects, the condition in mouse models deviates considerably from AD in humans. Murine models with an elevated aggregation of Aβ show cognitive impairment before Aβ pathology begins (Janus and Welzl, 2016). Furthermore, most mouse models do not replicate the dysfunction of tau proteins, which is another pathological hallmark of AD. Mice expressing physiological levels of both human APP and human tau might provide better models of the disease, since sporadic AD is a multifactorial disorder involving different cytotoxic mechanisms (Medina and Avila, 2014). Tau-based strategies have only recently come into focus in animal research, and a triple transgenic AD mouse model harboring transgenes of mutants of APP, PSEN1 and Tau has been developed (Oddo et al., 2003).
In contrast to face validity, construct validity (sound theoretical rationale of the animal model based on good understanding of the disease, e.g. same pathomechanisms in models and humans) and predictive validity (translation of findings in models to humans, e.g. discrimination between successful and unsuccessful therapies of the disease) remain problematic (Bilkei-Gorzo, 2014; Willner, 1986). In short, the available AD models have, at best, face validity, with the animal analogue merely sharing some phenomenological similarities with the disease.
The frequent failure to translate successful therapy from preclinical to human studies in AD research (Cummings et al., 2014; Cummings, 2021) may be the result of too limited a mirroring of human pathology in animal models (Banik et al., 2015). The main goal in the development of animal models of AD is to simulate its neuropathological characteristics and to correlate them to cognitive functions. However, whether the pathological changes have similar biological consequences in animal models and humans remains unknown. While some morphological similarities exist between brain alterations in AD and transgenic mouse models, there is a difference in biochemical composition of deposited Aβ (Kalback et al., 2002). Moreover, on the basis of observations in transgenic rats and mice, the causal relationship between amyloid plaques and cognitive impairment has been called into question. The Tg6590 rat model expresses the Swedish mutation of APP and does not show mature plaque formation (Kloskowska et al., 2010), while the Tg2576 mouse equivalent displays substantial plaques (Hsiao et al., 1996). Interestingly, cognitive deficits have been found in both models.
Another limitation of transgenic mouse models is that the progressive neuronal loss found in the hippocampus and specific neocortical regions in human AD (Gómez-Isla et al., 1996) is not replicated in most models. Furthermore, cognitive impairment occurs at different stages of the pathogenetic process in rodent models and humans, with impairment of cognitive functions present in animals prior to or at the onset of plaque development and in humans many decades following the development of plaques (Drummond and Wisniewski, 2017).
AD is not a homogeneous disease entity (Korczyn, 2013) and is likely to be caused by multiple factors, including many modifiable ones, with the action of varying combinations of these factors in different individuals leading to the development of AD. Transgenic murine models represent only those individuals with familial AD, which constitute less than 1% of all people with AD. No existing murine model is capable of fully reproducing the features of disease progression characteristic of the vast majority of AD cases, i.e. sporadic, late-onset AD. Different etiopathogenetic processes may lead to the same pathological phenotype. Due to cognitive decline and the similarity of the neuropathological changes, the genetic and sporadic cases of AD are (incorrectly) combined to a single disease entity. However, genetic and sporadic cases of AD result from different causes and do not therefore form a single nosological entity with a common etiopathogenesis. Amyloid deposition may be the result of various contributing factors, such as trauma or hypoxia (Gatson et al., 2013; Huang et al., 2012; Lin, 2013), and does not therefore constitute a specific etiopathogenesis (Korczyn, 2008, 2013; Robakis, 2014).
Since various clinical trials assessing compounds targeting amyloid have failed to reach the primary clinical end-points (Huang et al., 2020; Mehta et al., 2017), it appears doubtful whether strategies targeting a decrease in production of or removal of Aβ are useful in AD treatment. A major translational issue is the discrepancy between the timeline of disease development in murine models versus humans (Myers and McGonigle, 2019). Amyloid plaques can appear in relatively young mice only a few months after birth, and this difference may partly explain why drugs that are successful in animals are ineffective in humans. At best, mouse models might reflect early stages of AD long before a diagnosis is made. The clinical trials of drugs targeting Aβ may have failed because the treatments are introduced too late to halt neuronal death. Whether or not anti-Aβ-based treatment strategies can provide clinical benefits is questionable, and any potential benefits may be limited to familial AD, with its prominent amyloid pathogenesis. Furthermore, treatment would probably need to be introduced early in the course of the pathological process in order to prevent neurodegeneration.
| 3. Future directions | ▴Top |
Transgenic mouse models have been valuable tools in elucidating pathogenetic mechanisms that may be relevant to human AD (Janus and Welzl, 2016). For example, they have provided valuable information on the importance of oxidative stress, inflammation and mitochondrial dysfunction in the pathogenesis of AD (Du et al., 2008; Lee et al., 2010). However, the utility of transgenic models in the development of novel treatment strategies and as preclinical testbeds of new drugs has been less than satisfactory. For many years, no new drugs effective in the treatment of AD have been approved (Berk and Sabbagh, 2013; Cummings et al., 2014; Cummings, 2021). The premature translation to humans of highly successful reductions of certain pathological changes in animal models that insufficiently mirror the neurobiology and symptomatology of AD may be a cause of the failure to develop effective treatments (Banik et al., 2015; Cummings et al., 2014). Over 99% of potential AD drugs tested successfully in animal experiments have failed in human trials (Cummings et al., 2014). For example, Tg2576 is a model strain based on a single familial AD transgene, and the relationship between amyloid pathology and impaired cognitive performance has been confirmed in this transgenic model. While Tg2576 mice have shown improved pathology and cognition following the administration of hundreds of agents in preclinical experiments (Zahs and Ashe, 2010), none of these compounds have shown beneficial clinical effects in people with AD. Taken together, the results of clinical trials in humans do not match those of animal experiments. It has therefore been suggested that research focus more on research in human patients (Horrobin, 2003).
Interventions using drugs or food bioactives need to ensure that these treatments are administered in sufficient doses over long enough periods of time. In view of the need to start trials in preclinical stages of AD before the appearance of manifest symptoms, this may be problematic. In regard to food science, interactive effects of nutrients on brain health may be more significant than those of single agents. In the prospective Oregon Brain Aging Study, various plasma biomarkers related to diet were analyzed in older participants with a mean age of 87 years (Bowman et al., 2012). This analysis yielded three distinct patterns relevant for age-related cognitive functioning and MRI measures. Two of these patterns were positively associated with cognitive and MRI outcomes: (1) a pattern high in plasma vitamins B, C, D and E and (2) a pattern high in omega-3 fatty acids. In contrast, the third pattern, which was high in trans-fatty acids, was associated with worse cognitive performance and a greater reduction in brain volume (Bowman et al., 2012). These findings emphasize the role of nutritional patterns in brain health, and the role of food bioactives in age-related cognitive decline should be investigated in combination with other dietary factors.
Since AD unfolds over several decades, trial design poses a great challenge. Studies attempting to test disease-modifying approaches in AD face the problem that neuropathological alterations begin to develop many years prior to the appearance of clinical symptoms. Major challenges of AD prevention trials include the need for large samples of community-dwelling people, long follow-up durations, an optimum time window for intervention and clinically relevant outcomes (Whiteley et al., 2020). The identification of populations at increased risk of AD on the basis of age and genetic or clinical factors will be important, as will the need to administer preventive treatments to cognitively unimpaired elderly people before the disease is fully expressed. Without valid biomarkers of subclinical AD, interventions in human trials cannot be introduced before clinical symptoms appear, which, considering the extended pathogenesis of AD over many years, may be too late. Furthermore, due to the heterogeneous etiology of AD with its variant genetic, environmental and lifestyle risk factors, specific biomarkers for different etiologies would be needed. The lack of biomarkers does not allow patient stratification based on different underlying pathologies.
| 4. Conclusion | ▴Top |
The available animal models of AD have so far been unsuccessful in identifying effective therapies. While some aspects of brain aging in humans can be found in aged non-human primates, AD and other age-related neurodegenerative disorders are characterized largely by pathological and clinical phenotypes that are specific to humans. Transgenic murine models reproducing the cause of rare, familial, early-onset AD have provided new insights into disease mechanisms, but they do not represent the common sporadic, late-onset AD forms of unknown etiology. While animal models should ideally replicate symptomatology, etiology and progression of a disease, transgenic mice mirror only limited aspects of the complex neurobiology of AD in humans. The use of these animals is unlikely to be sufficient to develop new therapies. In view of the heterogeneity of AD, a multi-target approach will probably be necessary.
Food scientists should be aware that the available AD animal models lack the ability to predict clinical success in humans. Thus, in view of the fruitless translation of preclinical findings to clinical practice, the discontinuation of the use of the available animal models of AD should be considered. Ultimately, hypotheses generated by animal experiments require confirmation in humans with AD, using pharmacological intervention and clinical trials. These trials need to distinguish between strategies enhancing cognition and modifying/preventing disease. Food bioactive research on AD should shift its focus and available resources from molecular studies in animals to human-centric, preventive approaches in clinically meaningful settings.
| References | ▴Top |